Regulatory remodeling in the allo-tetraploid frog Xenopus laevis
Elurbe DM, Paranjpe SS, Georgiou G, van Kruijsbergen I, Bogdanovic O, Gibeaux R, Heald R, Lister R, Huynen MA, van Heeringen SJ, Veenstra GJC.
Click here to view this article at Genome Biology.
Click here to view this article on Pubmed.
Click here to view this article on Xenbase.
In this study Elurbe et al. identify non-allelic homologous recombination between old repeats as the likely source of deletions that happen much more frequently on the S subgenome in X.laevis. Their analyses also show that new (i.e. subgenome-specific) enhancer elements in the L and S subgenomes of X.laevis originate from relatively young DNA transposons. Moreover, in experimental X.laevis egg x X.tropicalis sperm hybrid embryos, young species-specific DNA transposons gain enhancer properties specifically on the paternal chromosomes.
Abstract
Genome duplication has played a pivotal role in the evolution of many eukaryotic lineages, including the vertebrates. A relatively recent vertebrate genome duplication is that in Xenopus laevis, which resulted from the hybridization of two closely related species about 17 million years ago. However, little is known about the consequences of this duplication at the level of the genome, the epigenome, and gene expression. The X. laevis genome consists of two subgenomes, referred to as L (long chromosomes) and S (short chromosomes), that originated from distinct diploid progenitors. Of the parental subgenomes, S chromosomes have degraded faster than L chromosomes from the point of genome duplication until the present day. Deletions appear to have the largest effect on pseudogene formation and loss of regulatory regions. Deleted regions are enriched for long DNA repeats and the flanking regions have high alignment scores, suggesting that non-allelic homologous recombination has played a significant role in the loss of DNA. To assess innovations in the X. laevis subgenomes we examined p300-bound enhancer peaks that are unique to one subgenome and absent from X. tropicalis. A large majority of new enhancers comprise transposable elements. Finally, to dissect early and late events following interspecific hybridization, we examined the epigenome and the enhancer landscape in X. tropicalis × X. laevis hybrid embryos. Strikingly, young X. tropicalis DNA transposons are derepressed and recruit p300 in hybrid embryos. The results show that erosion of X. laevis genes and functional regulatory elements is associated with repeats and non-allelic homologous recombination and furthermore that young repeats have also contributed to the p300-bound regulatory landscape following hybridization and whole-genome duplication.

Fig. 1. Alignment of a region on chromosome 8 in X. tropicalis and the X. laevis L and S subgenomes annotated with experimental ChIP-seq data (gastrula-stage embryos; NF stage 10.5). Shown are the gene annotation (black), repeats (gray), ChIP-seq profiles for H3K4me3 (green), p300 (yellow), RNA Polymerase II (RNAPII; brown), and H3K36me3 (dark green). The sequence conservation is indicated by gray lines. Conserved H3K4me3 and p300 peaks are denoted by green and yellow lines, respectively. The anp32e gene is expressed in X. tropicalis and both the L and S subgenome of X. laevis. The plekho1 gene, on the other hand, has lost promoter and enhancer activity on the X. laevis S locus and shows no experimental evidence of being expressed.
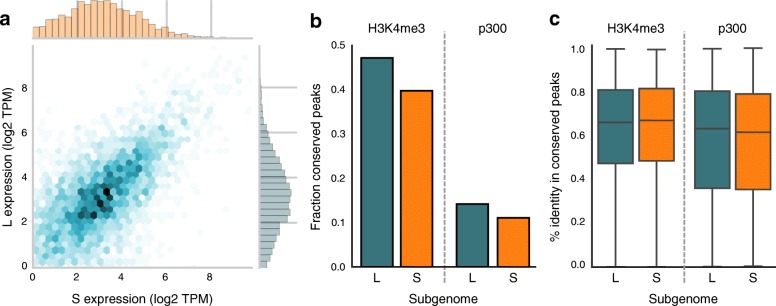
Fig. 2. a Scatterplot of the expression level (log2 TPM) of L and S homeologs that are both expressed. The expression level of homeolog genes is generally similar (Pearson R = 0.60, p < 1e-300). b Fraction of epigenetic signals (“peaks”) conserved in X. laevis compared to X. tropicalis. Promoters appear more conserved than enhancers; S has lost more epigenetic elements than L. c Active functional elements are equally conserved between L and S compared to X. tropicalis. The background level of sequence conservation in fourfold degenerate sites from coding sequences with respect to X. tropicalis is 78.4% in L and 77.7% in S.

Fig. 3. The S subgenome has more and larger deletions than L. a Size frequency distribution of deletions (top) and size ratio of LΔS deletions relative to SΔL deletions as a function of deletion size (bottom). b An example of a gene (grlx2) that has lost the promoter on the S genome due to a deletion. Shown are the gene annotation (black), ChIP-seq profiles for H3K4me3 (green), RNAPII (brown), and H3K36me3 (dark green). The sequence conservation is indicated by gray lines. c The log2 fold difference between the observed number of deleted basepairs and the expected number (mean of 1000 randomizations). The fold difference is calculated per chromosome and summarized in a boxplot. Intergenic 1 kb distance from a gene, Intronic introns, Exonic UTRs + CDS, IntronicTx introns from genes actively transcribed, ExonicTx exons from genes actively transcribed, p300 genomic fragments having a p300 peak, H3K4me3 genomic fragments having a H3K4me3 peak. The asterisks mark significant differences between the L and S chromosomes (p < 0.001, Mann–Whitney U test). d Retained regions associated with deletions are enriched for relatively long repeats (p < 1e-52 for both LΔS and SΔL; Mann–Whitney U test). e 1 kb flanks of the retained regions are more similar to each other than random genomic regions of the same size (p < 1e-114 and 1e-83 for LΔS and SΔL, respectively; Mann–Whitney U test).
Adapted with permission from BMC Genome Biology: Elurbe et al. (2017). Regulatory remodeling in the allo-tetraploid frog Xenopus laevis. Genome Biol. 2017 Oct 24;18(1):198. doi: 10.1186/s13059-017-1335-7. Copyright 2017.
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/