Single Amino Acid Change Underlies Distinct Roles of H2A.Z Subtypes in Human Syndrome
Cell Volume 178, Issue 6, 5 September 2019, Pages 1421-1436.e24, https://doi.org/10.1016/j.cell.2019.08.002
Rachel S. Greenberg, Hannah K. Long, Tomek Swigut, Joanna Wysocka
Highlights
• FHS-associated truncations of SRCAP AT-hooks lead to loss of nuclear localization
• H2A.Z.2 knockdown mimicks and overexpression rescues FHS craniofacial phenotypes
• H2A.Z.1 and H2A.Z.2 show subtle preferences in incorporation at promoters vs enhancers
• AT-rich enhancers and their associated genes are sensitized to FHS SRCAP truncations
Click here to view article at Cell.
Click here to view article on Pubmed.
Click here to view article on Xenbase (figures and supplemental figures).
Abstract
The developmental disorder Floating-Harbor syndrome (FHS) is caused by heterozygous truncating mutations in SRCAP, a gene encoding a chromatin remodeler mediating incorporation of histone variant H2A.Z. Here, we demonstrate that FHS-associated mutations result in loss of SRCAP nuclear localization, alter neural crest gene programs in human in vitro models and Xenopus embryos, and cause craniofacial defects. These defects are mediated by one of two H2A.Z subtypes, H2A.Z.2, whose knockdown mimics and whose overexpression rescues the FHS phenotype. Selective rescue by H2A.Z.2 is conferred by one of the three amino acid differences between the H2A.Z subtypes, S38/T38. We further show that H2A.Z.1 and H2A.Z.2 genomic occupancy patterns are qualitatively similar, but quantitatively distinct, and H2A.Z.2 incorporation at AT-rich enhancers and expression of their associated genes are both sensitized to SRCAP truncations. Altogether, our results illuminate the mechanism underlying a human syndrome and uncover selective functions of H2A.Z subtypes during development.
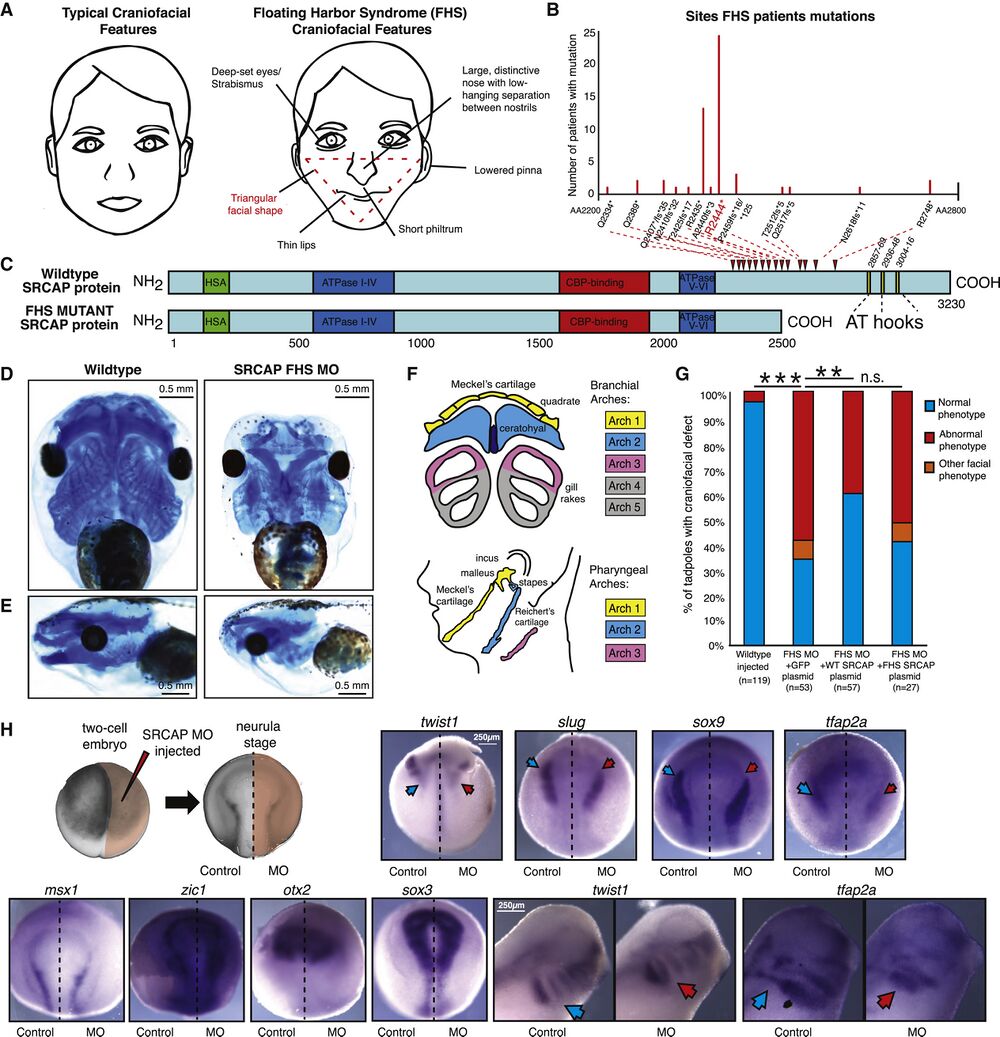
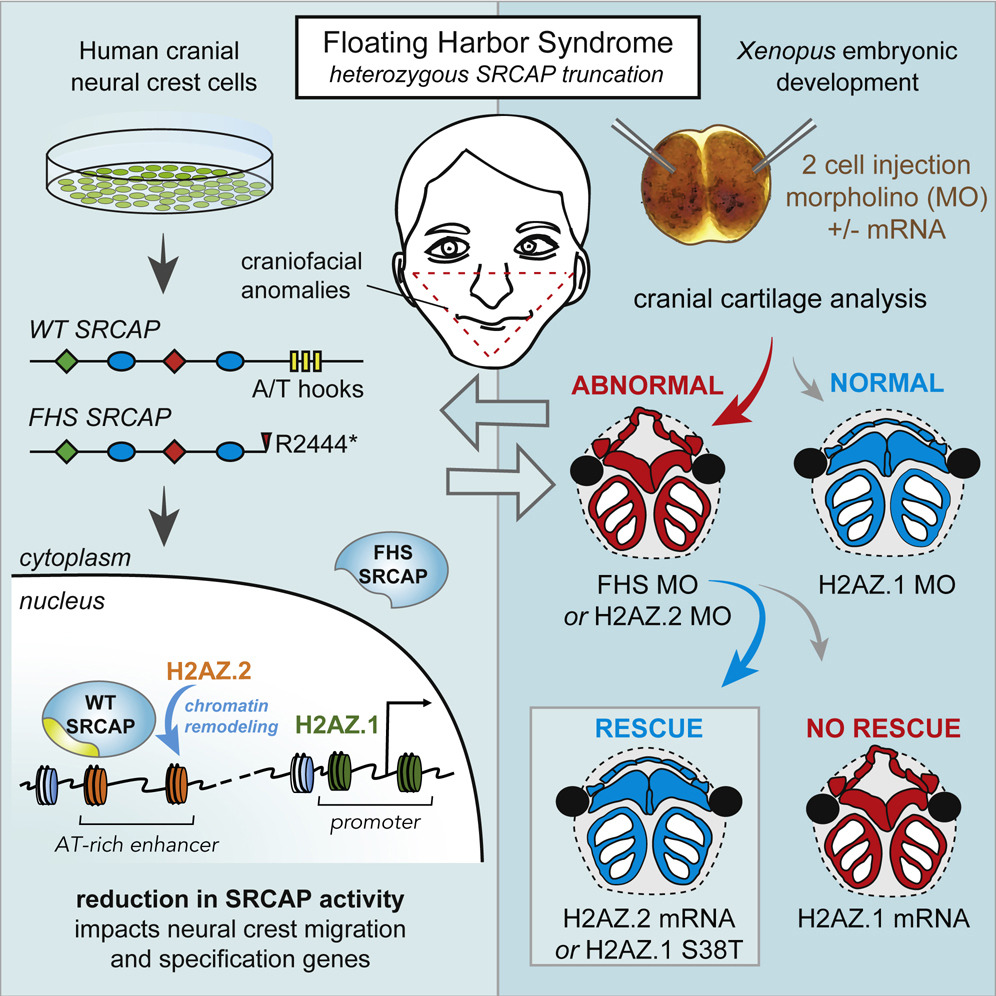
Figure 1. Recapitulating Floating-Harbor Syndrome SRCAP truncations in Xenopus laevis Affects Neural Crest Derived Craniofacial Structures. (A) Graphical representation of typical (left) and Floating-Harbor syndrome (right) craniofacial features. Characteristic triangular facial shape (most characteristic and diagnostic feature) demarcated in red. (B) Plot of frequencies of SRCAP mutations in Floating-Harbor syndrome probands. The x axis goes from amino acid 2200 to 2800, and each mutation is denoted on this axis. The most frequent mutation R2444∗ is highlighted in red. (C) Schematic of WT and the FHS truncated SRCAP proteins. The hot spot for FHS truncating mutations is indicated by red arrowheads. Protein domains are annotated with HSA in green, ATPase in blue, CBP-binding in red, AT-hooks in yellow. The amino acid scale is below the schematic. (D and E) Ventral (D) and side (E) view of X. laevis head with craniofacial cartilage stained with Alcian blue at stage 40, WT (mock-injected) and SRCAP FHS morphant (5.0 μM MO). Scale bar, 0.5 mm. Animals from n = 3 biologically independent experiments. (F) Diagram of homology between branchial arch structures in X. laevis to pharyngeal arches of the developing human face, with key homologous structures highlighted in matching colors. (G) Blinded quantification of rescue of characteristic craniofacial phenotype with co-injection of FHS MO and 200 pg pB CAG GFP-FLAG, or pB CAG WT-SRCAP-GFP-FLAG, or pB CAG FHS-SRCAP-GFP-FLAG. Statistical test used was Fisher’s exact test (FET). FET p value < 0.005 = ∗∗, FET p value < 10e−5 = ∗∗, FET p value > 0.05 = n.s. Animals from n = 4 independent experiments. (H) Diagram of injection set up at two-cell stage and of asymmetrical FHS SRCAP MO expression at neurula stage. In situ hybridization at neurula stage for neural crest specification genes twist1, slug, and sox9 (abnormal phenotype in 9/11, 5/5, 5/6 embryos, respectively), for neural crest induction and specification gene tfap2a (abnormal phenotype in 11/12 embryos), for neural plate border maintenance genes zic1 and msx1 (abnormal phenotype in 1/10, 0/6 embryos, respectively), for early neural patterning gene otx2 and for neural plate gene sox3 (abnormal phenotype in 0/5, 1/5 embryos, respectively), with 5.0 μM FHS MO injected on right side only, control on left. Ventral side shown, with anterior at top and posterior at bottom. Scale bar, 250 μm for neurula images. In situ hybridization at tailbud stage (stage 28), with each pair of images from same animal (control image flipped in vertical plane). In situ probes twist1 and tfap2a (abnormal phenotype in 8/10 and 11/13 embryos, respectively) visualize neural crest migration. Scale bar, 250 μm for tailbud images. Blue arrows denote normal gene expression pattern, red arrows denote impact on expression for FHS morphant. Image brightness and color adjusted to optimize visualization. See also Figure S1, Table S1, and Video S1.
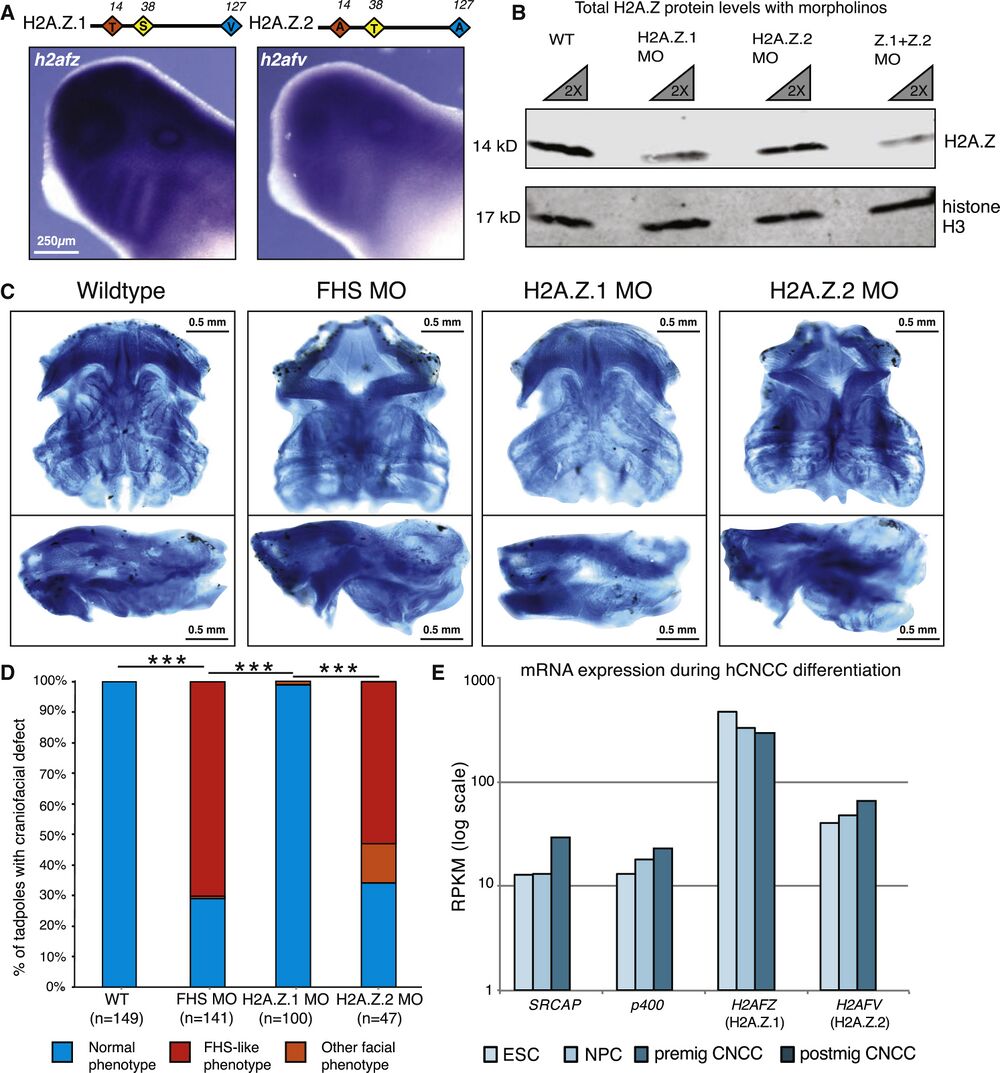
Figure 4. Knockdown of H2A.Z.2 Phenocopies the Craniofacial Features of FHS Morphant Frogs. (A) Schematic of H2A.Z.1 and H2A.Z.2 proteins. Red, yellow, and blue diamonds denote three amino acids divergent between H2A.Z.1 and H2A.Z.2. In situ hybridization staining for h2afz and h2afv mRNA at tailbud stage in WT X. laevis tadpoles. Scale bar, 250 μm. (B) Western blot of cellular extract from dissected X. laevis at tailbud stage, with WT and 2.5 μM H2AFZ MO and 2.5 μM H2AFV MO samples used. Antibodies against total-H2A.Z and total histone H3 (loading control). 1× and 2× dilution of each sample. Imaged and quantified on LI-COR Odyssey (see Figure S2B). (C) Ventral and side views of dissected X. laevis cartilage stained with Alcian blue at stage 40, WT (mock injected), SRCAP FHS morphant (SRCAP truncation) (5 μM), H2A.Z.1 morpholino (2.5 μM morpholino), and H2A.Z.2 morpholino (2.5 μM morpholino). Scale bar, 0.5 mm. Animals from n = 3 biologically independent experiments. (D) Blinded quantification of characteristic craniofacial phenotype. Statistical test was Pearson's chi-squared 2-sample test for equality of proportions with continuity correction. ∗∗∗p value <2.2e−16. Animals from n = 3 independent experiments. (E) mRNA expression of indicated genes (measured by reads per kilobase of transcript per million [RPKM]) from hESCs, neural precursor cells (NPCs), pre-migratory neural crest cells (premig NCCs), and post-migratory neural crest cells (postmig NCCs) (Rada-Iglesias et al., 2012). See also Figure S4.
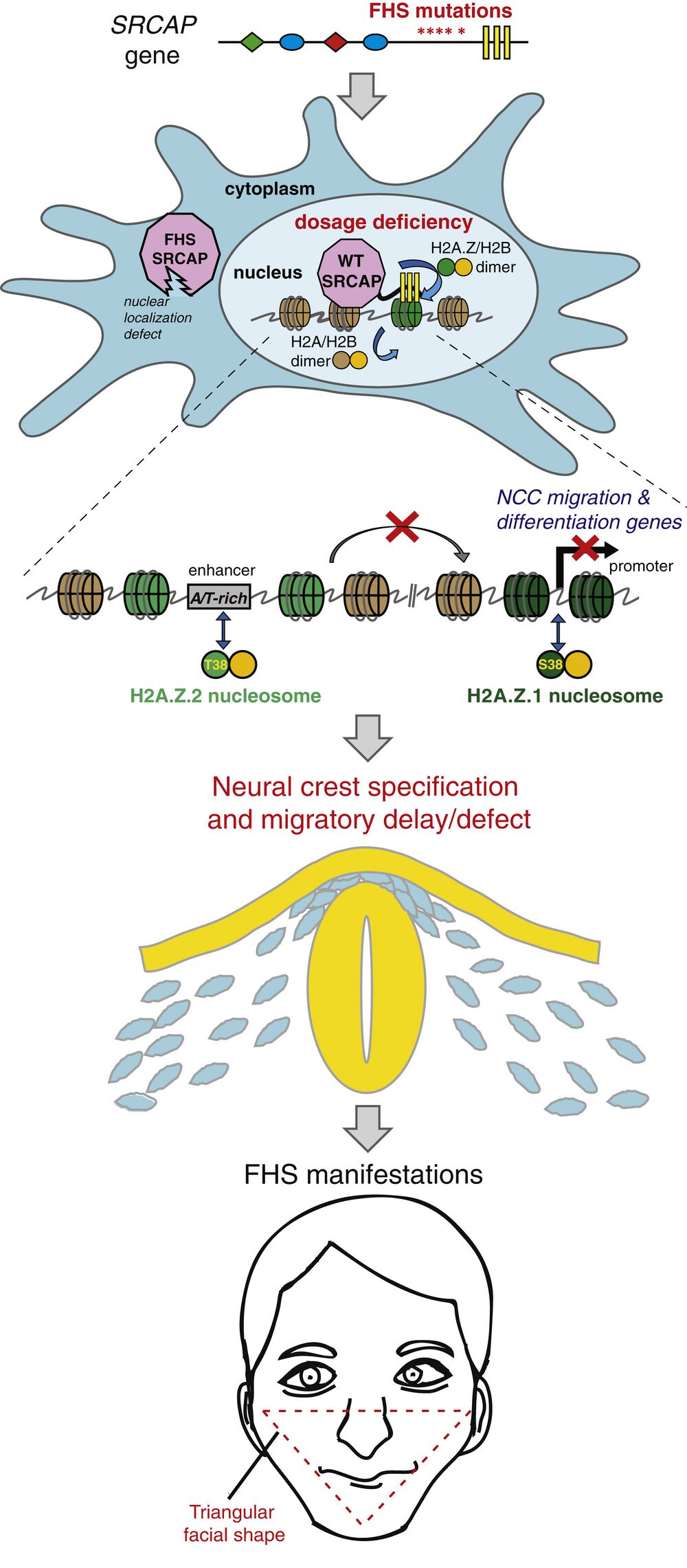
Figure 7. Proposed Model of Floating-Harbor Syndrome and H2A.Z Subtype Specialization. In FHS, heterozygous SRCAP mutation truncates the protein prior to DNA-binding AT-hooks, causing loss of SRCAP from nucleus and chromatin. With a diminished dose of functional SRCAP present in FHS, nuclear H2A.Z-remodeling activity is reduced. Genomic incorporation pattern of the two H2A.Z subtypes is qualitatively similar, but is biased toward promoters for H2A.Z.1 and AT-rich enhancers for H2A.Z.2. In FHS CNCCs, H2A.Z.2 is preferentially lost from AT-rich enhancers, and associated genes are downregulated. These sensitized regions regulate genes important for CNCC migration and differentiation. FHS patients have craniofacial anomalies related to defects in these developmental processes. See also Figure S7.
Adapted with permission from Cell Press on behalf of Cell Greenberg et al. (2019). Single Amino Acid Change Underlies Distinct Roles of H2A.Z Subtypes in Human Syndrome. Cell Volume 178, Issue 6, 5 September 2019, Pages 1421-1436.e24. doi: 10.1016/j.cell.2019.08.002. Copyright (2019).
Last Updated: 2019-09-12