Epigenetic sperm programming
Epigenetic homogeneity in histone methylation underlies sperm programming for embryonic transcription
Mami Oikawa, Angela Simeone, Eva Hormanseder, Marta Teperek, Vincent Gaggioli, Alan O’Doherty, Emma Falk, Matthieu Sporniak, Clive D’Santos, Valar Nila Roamio Franklin, Kamal Kishore, Charles R. Bradshaw, Declan Keane, Thomas Freour, Laurent David, Adrian T. Grzybowski, Alexander J. Ruthenburg, John Gurdon & Jerome Jullien
Nature Communications volume 11, Article number: 3491 (2020)
Click here to view article at Nature Communications.
Click here to view article on Pubmed.
Click here to view article on Xenbase.
Abstract
Sperm contributes genetic and epigenetic information to the embryo to efficiently support development. However, the mechanism underlying such developmental competence remains elusive. Here, we investigated whether all sperm cells have a common epigenetic configuration that primes transcriptional program for embryonic development. Using calibrated ChIP-seq, we show that remodelling of histones during spermiogenesis results in the retention of methylated histone H3 at the same genomic location in most sperm cell. This homogeneously methylated fraction of histone H3 in the sperm genome is maintained during early embryonic replication. Such methylated histone fraction resisting post-fertilisation reprogramming marks developmental genes whose expression is perturbed upon experimental reduction of histone methylation. A similar homogeneously methylated histone H3 fraction is detected in human sperm. Altogether, we uncover a conserved mechanism of paternal epigenetic information transmission to the embryo through the homogeneous retention of methylated histone in a sperm cells population.
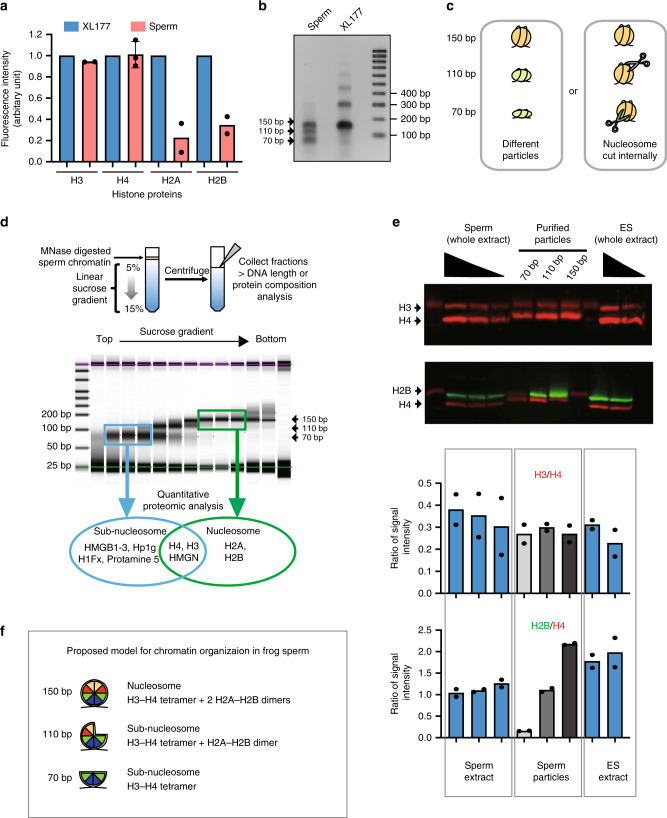
Fig. 1
Somatic level of histone H3/H4 is retained as nuclesosomes and subnucleosomes in sperm chromatin. a Xenopus laevis sperm core histones content relative to that found in a somatic cell (XL-177) as measured by quantitative WB (H2A, H2B, H3 n = 2, H4 n = 3, biologically independent samples error bar on H4 shows standard deviation). b DNA fragments generated by MNase digestion of Xenopus laevis sperm and somatic cell. c Schematic representation of the possible origin of subnucleosomal sized fragments generated by MNase treatment of sperm chromatin. d Nucleoproteic particles generated by MNase treatment of sperm are centrifugated on a sucrose gradient. Subsequently, particles isolated along the gradient are analysed for associated DNA fragment length (electrophoresis) and for associated proteins (mass spectrometry). e WB analysis confirms mass spectrometry analysis. Similar ratio of H3 to H4, and decreased level H2B to H4 are detected in subnucleosomes compared with nucleosomes. Xenopus sperm and mESCs are shown as control. Graphs below show the quantification of WB data (n = 2, biologically independent samples). f Model of core histone composition of Xenopus laevis sperm nucleosomal and subnucleosomal particle. Source data related to a, b, d and e are provided as Source Data files.
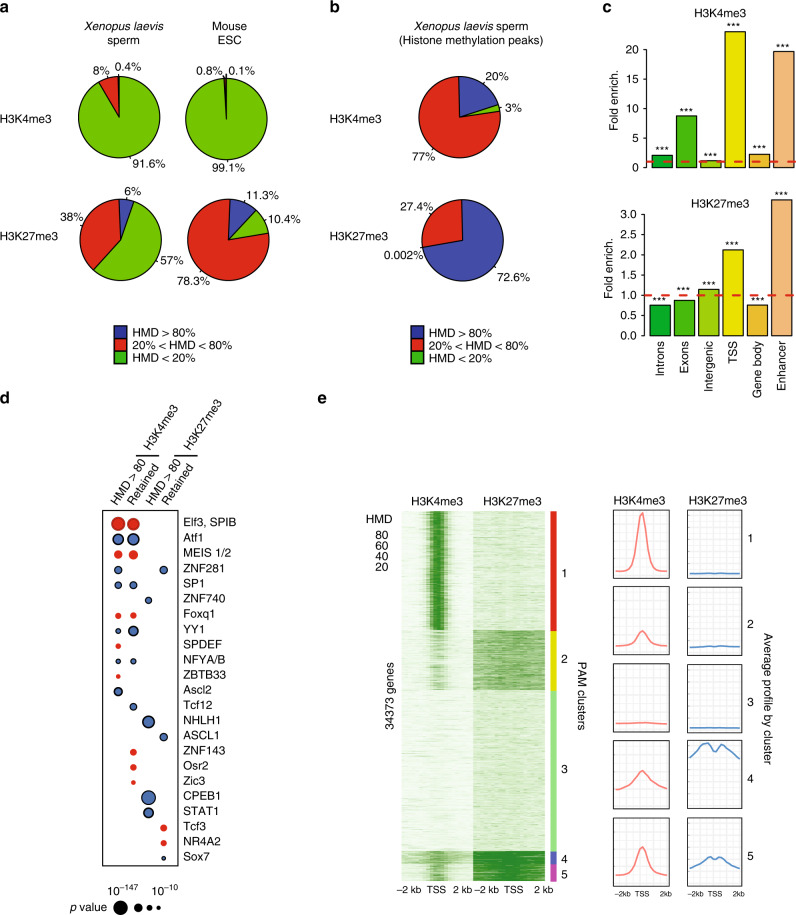
Fig. 3
A fraction of the genome harbours methylated H3K4 and/or H3K27 at the same location in most sperm of a population. a Percentage of the genome with different range of apparent histone H3 methylation density (HMD) on Lysine 4 and 27 in Xenopus Laevis sperm and mouse ESC. b Percentage of H3K4me3 and H3K27me3 peaks with different range of HMD in Xenopus Laevis sperm. c Fold enrichment (observed/random) over 1000 randomisations of peaks with homogeneous histone methylation (HMD > 80) at the indicated genomic features; ***: empirical p value < 1e−3. ICe-ChIP data from two independent replicates were pooled. d Dot matrix showing transcription factors with enriched motifs (y-axis) in the different histone methylation categories (x-axis). Circle size represents –log10 (p value) of the motif enrichment; and the circle colour indicates whether evidences exist indicating that the corresponding transcription factors is present maternally (blue) or not (red). Retained HMD > 80 peaks correspond to sperm histone methylation peaks maintained after extract treatment as in Fig. 5. e Heat map after PAM clustering of promoters (TSS +/−2 kb) according to histone H3-methylation density on Lysine 4 and 27. The plots on the left show the average HMD profile for each cluster. Source data related to a, b, d, and e are provided as Source Data files.
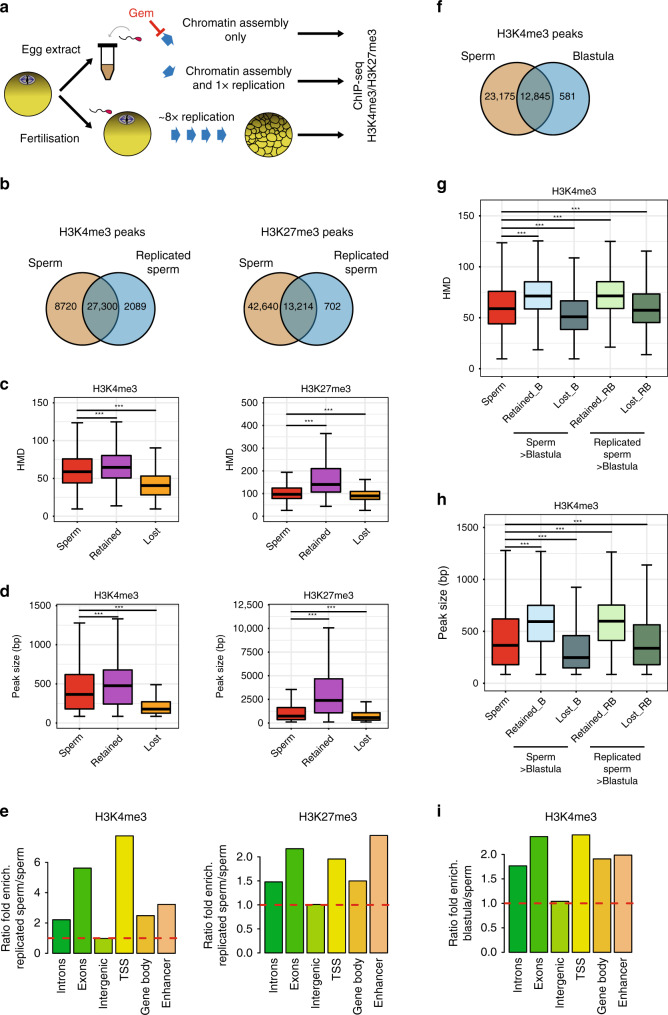
Fig. 5
Homogeneously methylated sperm histones are maintained during early embryonic replication. a Experimental setup to monitor the fate of sperm-methylated histone peaks after replication. b Overlap between peaks of H3K4me3 and H3K27me3 before and after replication of sperm chromatin in egg extract. c Boxplots of HMD for all sperm peaks and for peaks that are lost or retained after replication. d Boxplots of the size of all sperm peaks, and of peaks that are lost or retained after replication. Data in c and d are obtained from N. sperm peaks H3K4me3: 36020; N. sperm retained H3K4me3: 27300; N sperm lost H3K4me3: 8715. N. sperm peaks H3K27me3: 55854; N. sperm retained H3K27me3: 13214; N sperm lost H3K27me3: 42635. ***p value < 1e−3 (two-sample Kolmogorov–Smirnov test). e Ratio of fold enrichment of peaks retained after replication over those lost after replication at indicated genomic features. Fold enrichments (observed/random) were obtained from 1000 randomisations and all instances showed an empirical
p value < 1e−3. f Overlap between peaks of H3K4me3 in sperm and in blastula embryos. g Boxplots indicating HMD for all sperm peaks and for peaks that are lost or retained in blastula compared with sperm, and lost or retained in blastula compared with replicated sperm. h Boxplots of peak sizes for all sperm peaks and for peaks lost and retained in blastula compared with sperm, and in blastula compared with replicated sperm. In g and h ***p value < 1e−3 and are obtained by the two-sample Kolmogorov–Smirnov test. i Ratio of fold enrichment of peaks retained in blastula versus those in sperm at indicated genomic features. This ratio has been obtained as in e. HMD is from data pooled from two independent replicates. Peaks retention/lost are consensus from three independent replicates.
Adapted with permission from Springer Nature on behalf of Nature Communications: Oikawa et al. (2020). Epigenetic homogeneity in histone methylation underlies sperm programming for embryonic transcription. Nat Commun. 2020 Jul 13;11(1):3491. doi: 10.1038/s41467-020-17238-w.
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Last Updated: 2020-09-15