Biallelic variants in COPB1 cause a novel, severe intellectual disability syndrome with cataracts and variable microcephaly
Macken WL, Godwin A, Wheway G, Stals K, Nazlamova L, Ellard S, Alfares A, Aloraini T, AlSubaie L, Alfadhel M, Alajaji S, Wai HA, Self J, Douglas AGL, Kao AP, Guille M, Baralle D.
Genome Med February 25, 2021; 13 (1): 34.
Click here to view article at Genome Medicine.
Click here to view article at Pubmed.
Click here to view article on Xenbase.
Abstract
Background
Coat protein complex 1 (COPI) is integral in the sorting and retrograde trafficking of proteins and lipids from the Golgi apparatus to the endoplasmic reticulum (ER). In recent years, coat proteins have been implicated in human diseases known collectively as “coatopathies”.
Methods
Whole exome or genome sequencing of two families with a neuro-developmental syndrome, variable microcephaly and cataracts revealed biallelic variants in COPB1, which encodes the beta-subunit of COPI (β-COP). To investigate Family 1’s splice donor site variant, we undertook patient blood RNA studies and CRISPR/Cas9 modelling of this variant in a homologous region of the Xenopus tropicalis genome. To investigate Family 2’s missense variant, we studied cellular phenotypes of human retinal epithelium and embryonic kidney cell lines transfected with a COPB1 expression vector into which we had introduced Family 2’s mutation.
Results
We present a new recessive coatopathy typified by severe developmental delay and cataracts and variable microcephaly. A homozygous splice donor site variant in Family 1 results in two aberrant transcripts, one of which causes skipping of exon 8 in COPB1 pre-mRNA, and a 36 amino acid in-frame deletion, resulting in the loss of a motif at a small interaction interface between β-COP and β’-COP. Xenopus tropicalis animals with a homologous mutation, introduced by CRISPR/Cas9 genome editing, recapitulate features of the human syndrome including microcephaly and cataracts. In vitro modelling of the COPB1 c.1651T>G p.Phe551Val variant in Family 2 identifies defective Golgi to ER recycling of this mutant β-COP, with the mutant protein being retarded in the Golgi.
Conclusions
This adds to the growing body of evidence that COPI subunits are essential in brain development and human health and underlines the utility of exome and genome sequencing coupled with Xenopus tropicalis CRISPR/Cas modelling for the identification and characterisation of novel rare disease genes.
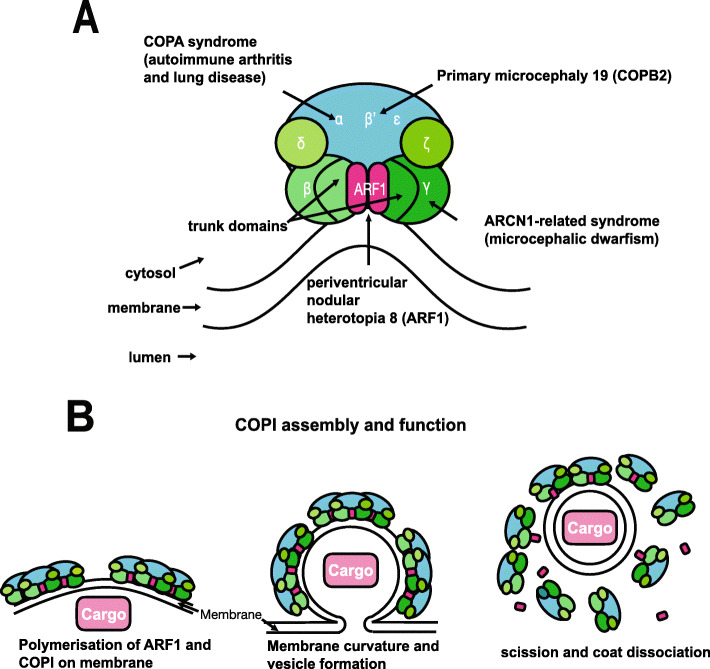
Fig. 1. Structure and function of the COP1 complex. a COPI consists of a scaffold “B-subcomplex” (blue) and an adaptor “F-subcomplex” (green). When GTP-bound, two ARF1 small GTPase molecules associate with the membrane and bind COPI via the β-COP and γ-COP subunits. A number of subunits of this complex have been implicated in human disease as shown. b COPI complexes and their ARF1 molecules associate into triads. Cargo, such as ER-resident proteins which need to be returned from the Golgi, are selected by binding directly with COPI subunits or indirectly through transmembrane receptors which in turn bind with COPI. COPI polymerises on the membrane enabling its deformation/curvature, and eventually budding and scission of the transport vesicle. When released, the vesicle’s coat is shed and ARF1 and COPI dissociate. Adapted from Nickel et al.
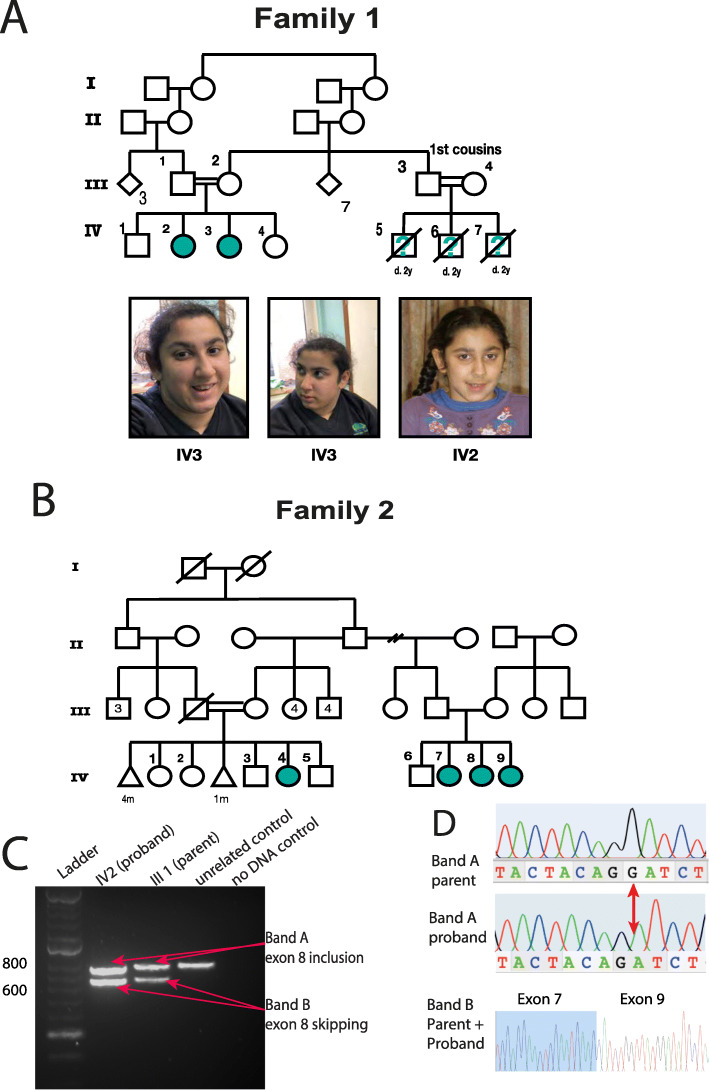
Fig. 2. Pedigrees and clinical presentation of individuals with COPB1 mutations. a Family 1 includes two affected individuals IV2 and IV3. Individuals IV5, IV6 and IV7 are also suspected to have been affected with microcephaly and developmental delay. They died in early childhood and no further details were available. IV2 and IV3 have minor dysmorphism with up-slanting palpebral fissures. b Family 2 includes four affected individuals from two nuclear families all from the same Saudi tribe. c Gel electrophoresis of RT PCR amplicons in Family 1 demonstrates 2 bands in the homozygous state (IV2, an affected individual) and in the heterozygous state (III1, an unaffected parent). d Electropherograms of band A and band B from the heterozygous parent and homozygous proband. In the proband’s trace, a G has effectively been deleted due to the creation of a new donor site by the G>T mutation. In band B, Exon 8 has been skipped (proband and parent).
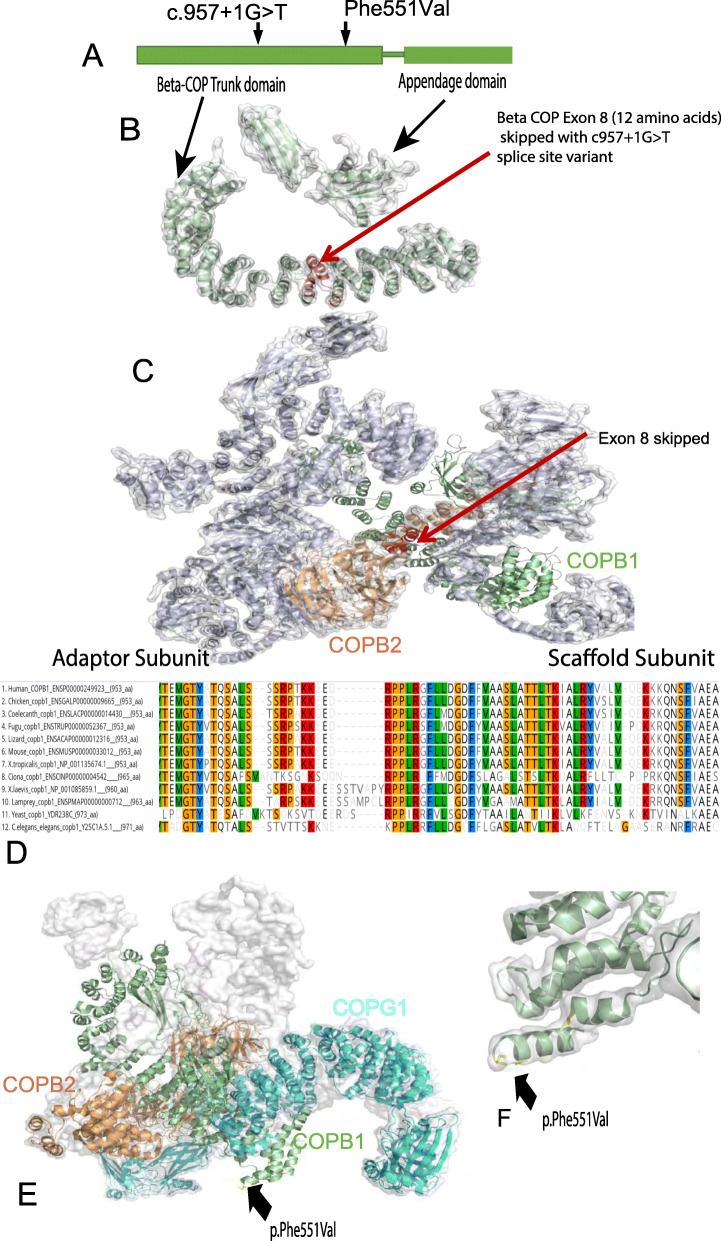
Fig. 3. Structure and conservation of COPB1 protein, and structural effect of missense variants. a Simple schematic diagram of COPB1 (β-COP) structure showing two main structural domains; trunk domain and appendage domain, and relative location of the c.957+1G>T and p.Phe551Val variants located towards the N terminal of the trunk domain. b COPB1 (β-COP) structure with the location of the 12 amino acids (in red) which are deleted due to exon 8 skipping caused by c957+1G>T. c COPB1 (β-COP) structure in context of COPI complex. Note exon 8 (red) makes up an important link between the scaffold and adaptor subcomplexes. d Alignment of COPB1 amino acid sequence from H. sapiens to yeast showing very high conservation of Phe551 across all species tested. e 3D structural modelling of COPB1 in complex with COPB2 and COPG1 showing the location of Phe551 near the site of interaction with COPG1. f Higher resolution image showing the location of Phe551Val mutation in a turn in the trunk domain.
Adapted with permission from Springer Nature on behalf of Genome Medicine: Macken et al. (2021). Biallelic variants in COPB1 cause a novel, severe intellectual disability syndrome with cataracts and variable microcephaly. Genome Med. 2021 Feb 25;13(1):34. doi: 10.1186/s13073-021-00850-w.20
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Last Updated: 2021-03-10