RAF1 deficiency causes a lethal syndrome that underscores RTK signaling during embryogenesis
EMBO Mol Med 2023 Apr 17;:e17078. doi: 10.15252/emmm.202217078.
Wong S, Tan YX , Loh AYT, Tan KY, Lee H, Aziz Z, Nelson SF, Özkan E, Kayserili H, Escande-Beillard N, Reversade B.
Click here to view article at EMBO Molecular Medicine.
Click here to view article on PubMed.
Click here to view article on Xenbase.
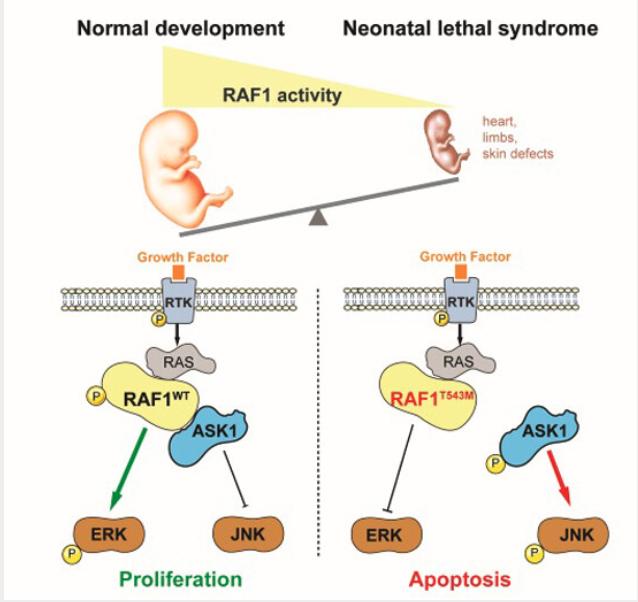
Synopsis
A novel recessive variant in RAF1 was identified as a possible cause of a hitherto unknown lethal neonatal syndrome with progeroid features. This private p.Thr543Met mutation abolished downstream ERK pathway signaling while increasing susceptibility to apoptosis via ASK1 de-repression.
- A homozygous p.Thr543Met mutation in RAF1 was found in a neonate presenting with a syndrome encompassing cutaneous, craniofacial, cardiac and limb anomalies.
- In cultured cells and in vivo using Xenopus assays the p.Thr543Met mutation behaved as a loss-of-function allele that abolishes downstream ERK pathway signaling in response to growth factor stimulation.
- p.Thr543Met knock-in in 293T cells revealed that RAF1T543M is unstable and more sensitive to stress-induced apoptosis via ASK1 de-repression.
- These findings describe, for the first time, the consequences of a loss of RAF1 function during human development, contrasting with gain-of-function mutations seen in cancer-prone RASopathies.
Abstract
Somatic and germline gain-of-function point mutations in RAF, one of the first oncogenes to be discovered in humans, delineate a group of tumor-prone syndromes known as the RASopathies. In this study, we document the first human phenotype resulting from the germline loss-of-function of the proto-oncogene RAF1 (a.k.a. CRAF). In a consanguineous family, we uncovered a homozygous p.Thr543Met variant segregating with a neonatal lethal syndrome with cutaneous, craniofacial, cardiac, and limb anomalies. Structure-based prediction and functional tests using human knock-in cells showed that threonine 543 is essential to: (i) ensure RAF1's stability and phosphorylation, (ii) maintain its kinase activity toward substrates of the MAPK pathway, and (iii) protect from stress-induced apoptosis mediated by ASK1. In Xenopus embryos, mutant RAF1T543M failed to phenocopy the effects of normal and overactive FGF/MAPK signaling, confirming its hypomorphic activity. Collectively, our data disclose the genetic and molecular etiology of a novel lethal syndrome with progeroid features, highlighting the importance of RTK signaling for human development and homeostasis.
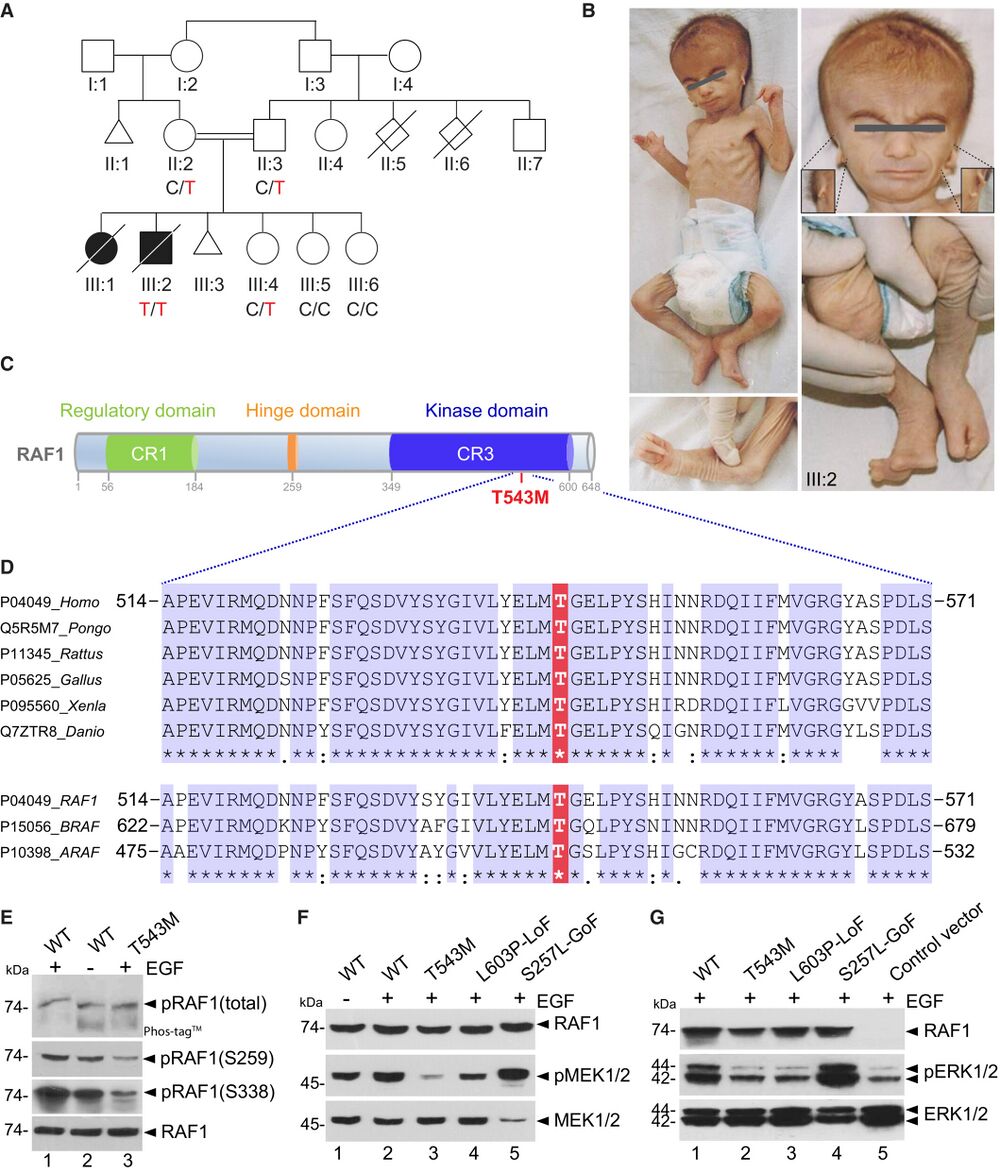
Figure 1. RAF1 germline homozygous missense mutation causes a neonatal lethal syndrome with progeroid features A. Pedigree of the Turkish consanguineous family reporting the neonatal progeroid syndrome. Square, male; circle, female; black shading, affected individuals; small triangle, aborted; double lines, consanguineous marriages; diagonal line, deceased. B. Pictures of affected proband (III:2) at 50 days showing general progeroid features. Note subcutaneous lipoatrophy, severe hypotonia, distinctive craniofacial features with absence of external ears (insets), cleft hands and feet with oligodactyly. C. Schematic of the RAF1 protein structure showing the three conserved regions (CRs). p.T543M is located in the catalytic kinase domain (CR3). D. Multiple sequence alignment showing conservation of T543 across species and among RAF family members. E. RAF1WT or RAF1T543M were transiently overexpressed in HEK293T cells for 48 h, followed by 16 h of serum starvation and EGF stimulation for 15 min. Phos-tag™ gel showing global levels of RAF1 phosphorylation and corresponding Western blot showing phosphorylation levels of Ser259 and Ser338 residues and total RAF1. F, G. All the RAF1 constructs were transiently overexpressed in HEK293T cells as described in (E). Western blotting showing (F) phosphorylated MEK1/2, total MEK1/2 and total RAF1 and (G) phosphorylated ERK1/2, total ERK1/2 and total RAF1. Source data are available.
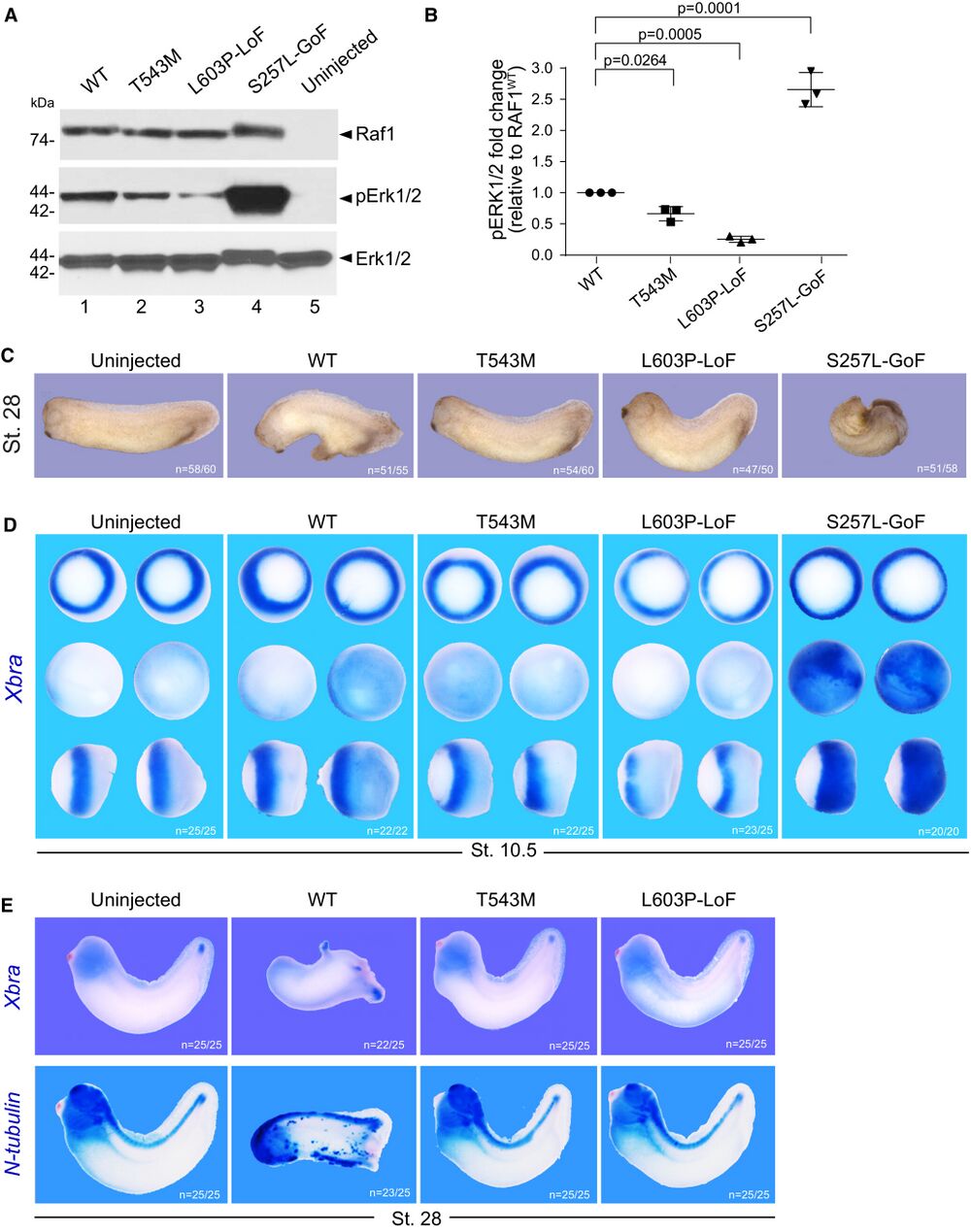
Figure 2. RAF1T543M allele behaves as a loss-of-function mutation in vivo A. Western blotting showing phosphorylated Erk1/2, total Erk1/2 and total Raf1 from Xenopus laevis embryos harvested at stage 28. B. Quantification of phosphorylated Erk1/2, normalized to WT Raf1 in three independent Western blots (Error bars indicate mean ± SEM. Ordinary one-way ANOVA). C. Representative images illustrating the effect of WT or mutants RAF1 mRNA injection into Xenopus embryos, at stage 28. D. WISH performed on stage 10.5 Xenopus embryos injected with WT or mutants RAF1 mRNA. Xbra (blue staining) marking mesoderm induction. Vegetal (top), animal (middle) and lateral (bottom) views. E. WISH performed on stage 28 Xenopus embryos injected with WT or mutants RAF1 mRNA. Blue staining reveals Xbra or N-tubulin markers depicting ectopic mesoderm induction, neural differentiation and brain posterization. Source data are available online for this figure.
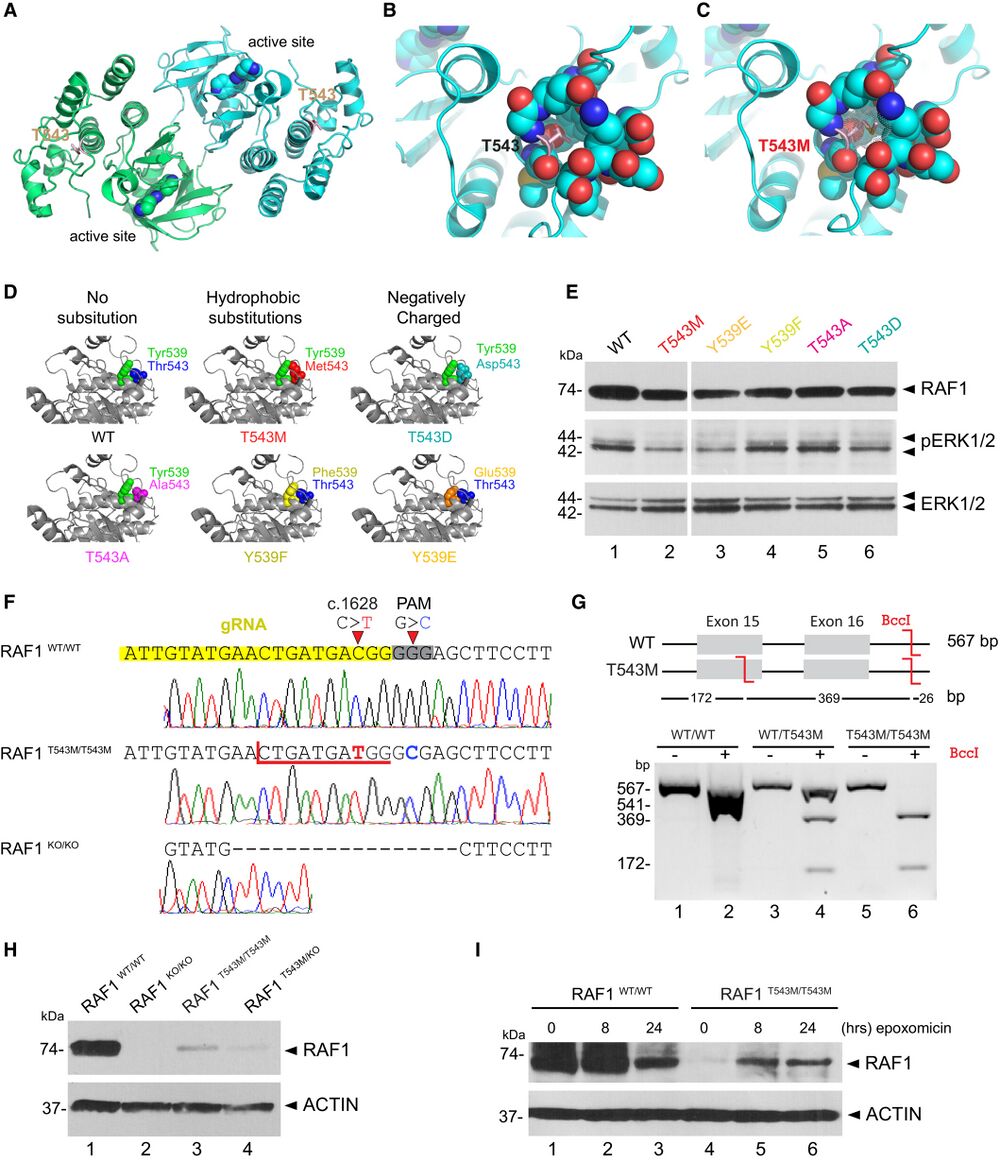
Figure 3. RAF1T543M/T543M knock-in cells reveal RAF1's compromised protein half-life A. RAF1 homodimer (PDB:3OMV) showing the location of Thr543 (drawn as ball-and-stick) on helical subdomain. Active site is occupied by an inhibitor drawn as spheres. B. Positioning of Thr543 in the pocket. C. Met543 in the pocket. The Met543 side chain clashes despite being modeled in the least sterically hindered rotamer. D. Predicted side chain interaction of Thr543 and Tyr539 variants. E. Western blot downstream signaling potential of structural mutants that induce steric (p.T543M) or electrostatic (p.Y539E or p.T543D) interference. F. RAF1T543M and RAF1KO/KO HEK293T cells were generated by CRISPR-Cas9. Top—bottom: Sanger-sequencing of RAF1 in WT HEK293T cells; a RAF1T543M clone showing the desired c.1628C>T (p.T543M) mutation and a synonymous PAM-disrupting c.1632G>C; a RAF1KO/KO clone with a homozygous 17 bp deletion. G. Confirmation of a clonal RAF1T543M line by restriction-digest of RAF1 exons 15–16 from gDNA of the genotypes shown with BccI. c.1628C>T introduces a BccI restriction site. The RAF1WT/T543M heterozygote genotype was obtained from the patient's father. H. Western Blot for endogenous RAF1 in HEK293T cell lines of genotypes indicated, at steady state. I. Western-blot showing that 8 and 24 h of epoxomicin treatment is sufficient to partially rescue RAF1 in RAF1T543M/T543M cells. ACTIN serves as loading control. Source data are available online for this figure.
Adapted with permission from EMBO Molecular Medicine on behalf of EMBO Press: Wong et al. (2023). RAF1 deficiency causes a lethal syndrome that underscores RTK signaling during embryogenesis. EMBO Mol Med 2023 Apr 17;:e17078. doi: 10.15252/emmm.202217078.
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
Last Updated: 2023-05-22