|
Literature for DOID 0050736: autosomal dominant disease
Xenbase Articles
(
| Functional and clinical characterization of a mutation in KCNJ2 associated with Andersen-Tawil syndrome., Lu CW,Lin JH,Rajawat YS,Jerng H,Rami TG,Sanchez X,DeFreitas G,Carabello B,DeMayo F,Kearney DL,Miller G,Li H,Pfaffinger PJ,Bowles NE,Khoury DS,Towbin JA, J Med Genet. August 1, 2006; 43(8):1468-6244. |
|
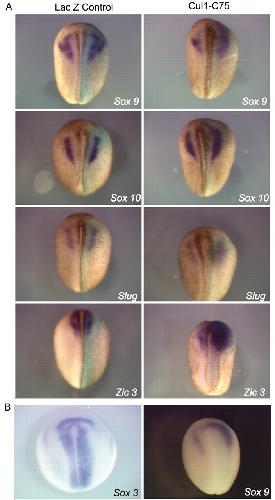
A dominant-negative form of the E3 ubiquitin ligase Cullin-1 disrupts the correct allocation of cell fate in the neural crest lineage.,
Voigt J,Papalopulu N,
Development. February 1, 2006; 133(3):1477-9129.
|
|
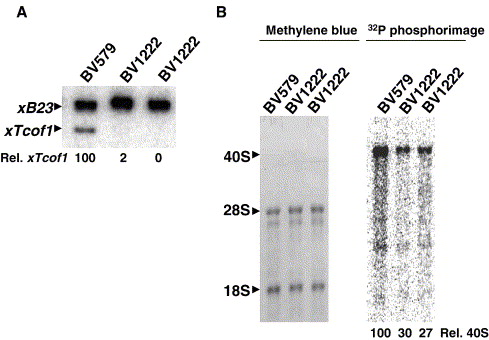
Cloning and functional characterization of the Xenopus orthologue of the Treacher Collins syndrome (TCOF1) gene product.,
Gonzales B,Yang H,Henning D,Valdez BC,
Gene. October 10, 2005; 359:1879-0038.
|
|
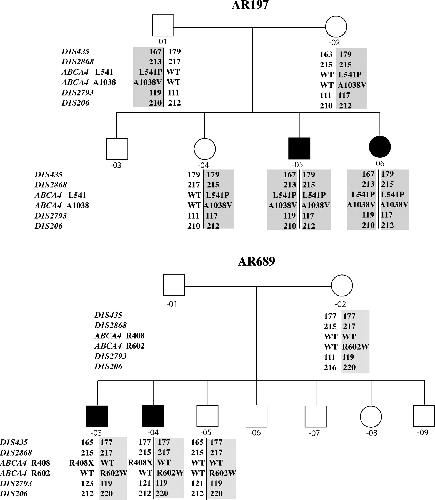
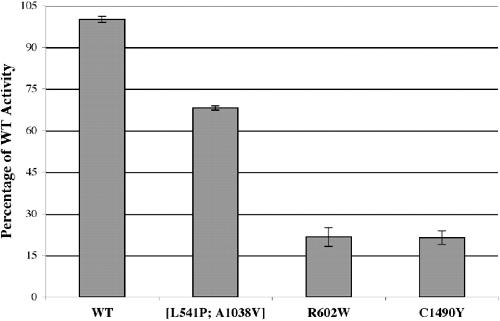
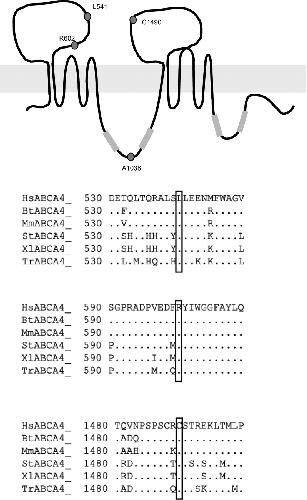
ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies.,
Wiszniewski W,Zaremba CM,Yatsenko AN,Jamrich M,Wensel TG,Lewis RA,Lupski JR,
Hum Mol Genet. October 1, 2005; 14(19):1460-2083.
|
| Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations., Splawski I,Timothy KW,Decher N,Kumar P,Sachse FB,Beggs AH,Sanguinetti MC,Keating MT, Proc Natl Acad Sci U S A. June 7, 2005; 102(23):1091-6490. |
|
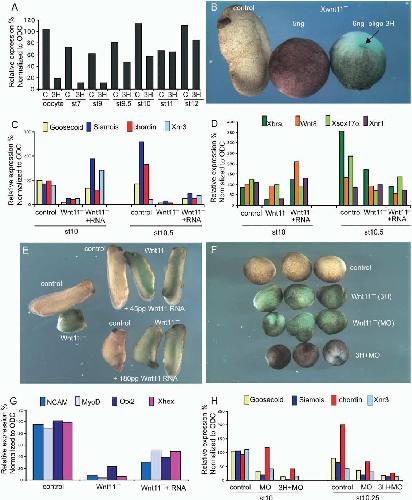
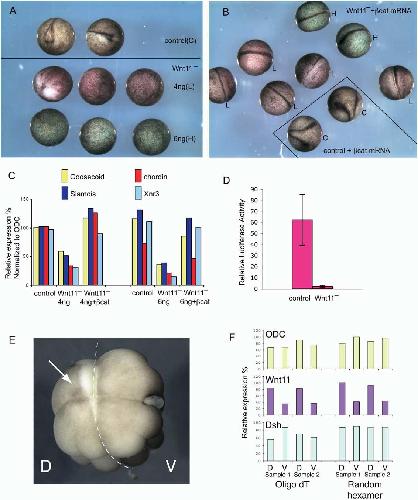
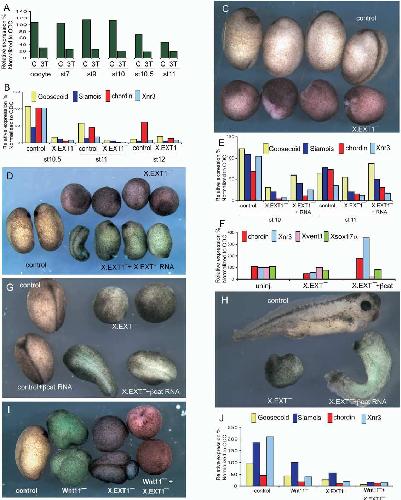
Maternal wnt11 activates the canonical wnt signaling pathway required for axis formation in Xenopus embryos.,
Tao Q,Tao Q,Yokota C,Puck H,Kofron M,Birsoy B,Yan D,Asashima M,Wylie CC,Lin X,Heasman J,
Cell. March 25, 2005; 120(6):1097-4172.
|
|
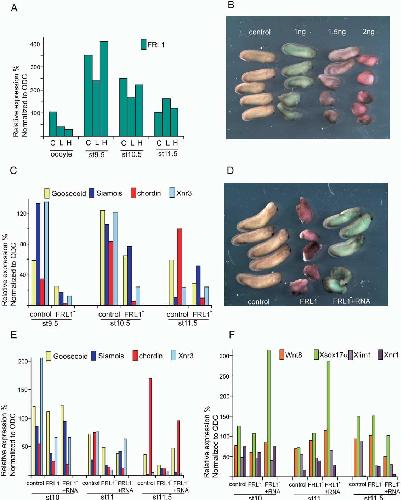
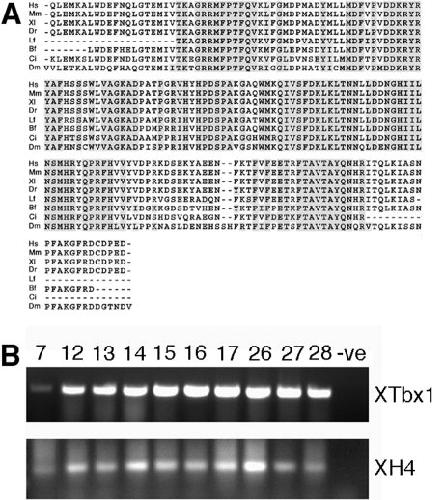
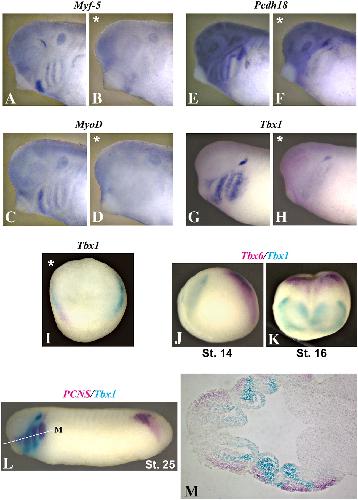
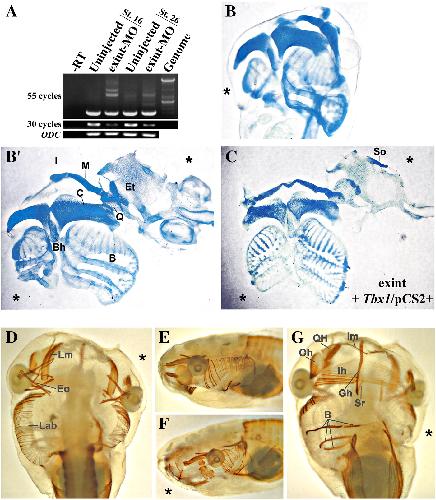
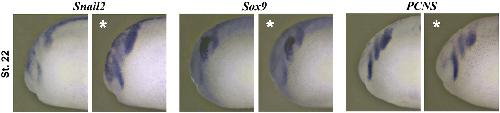
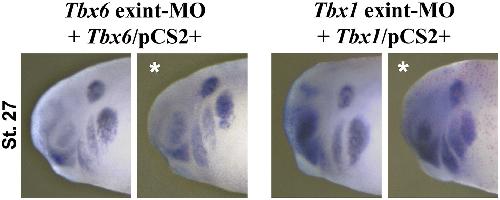
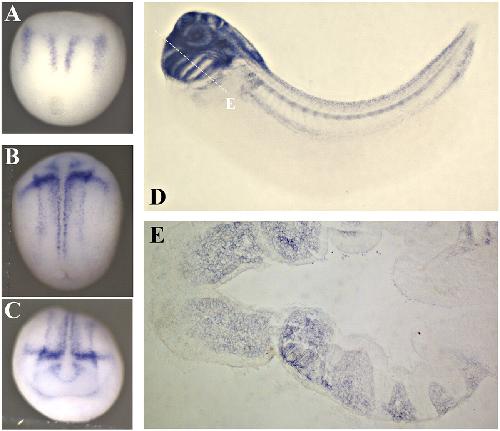
XTbx1 is a transcriptional activator involved in head and pharyngeal arch development in Xenopus laevis.,
Ataliotis P,Ivins S,Mohun TJ,Scambler PJ,
Dev Dyn. April 1, 2005; 232(4):1058-8388.
|
| Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism., Splawski I,Timothy KW,Sharpe LM,Decher N,Kumar P,Bloise R,Napolitano C,Schwartz PJ,Joseph RM,Condouris K,Tager-Flusberg H,Priori SG,Sanguinetti MC,Keating MT, Cell. October 1, 2004; 119(1):1097-4172. |
|
Mouse Zic5 deficiency results in neural tube defects and hypoplasia of cephalic neural crest derivatives.,
Inoue T,Hatayama M,Tohmonda T,Itohara S,Aruga J,Mikoshiba K,
Dev Biol. June 1, 2004; 270(1):1095-564X.
|
|
Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features.,
Roessler E,Du YZ,Mullor JL,Casas E,Allen WP,Gillessen-Kaesbach G,Roeder ER,Ming JE,Ruiz i Altaba A,Muenke M,
Proc Natl Acad Sci U S A. November 11, 2003; 100(23):1091-6490.
|
| Investigation of nuclear architecture with a domain-presenting expression system., Dreger CK,König AR,Spring H,Lichter P,Herrmann H, J Struct Biol. January 1, 2002; 140(1-3):1047-8477. |
| Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen's syndrome., Preisig-Müller R,Schlichthörl G,Goerge T,Heinen S,Brüggemann A,Rajan S,Derst C,Veh RW,Daut J, Proc Natl Acad Sci U S A. May 28, 2002; 99(11):1091-6490. |
| Pallister-Hall syndrome phenotype in mice mutant for Gli3., Böse J,Grotewold L,Rüther U, Hum Mol Genet. May 1, 2002; 11(9):1460-2083. |
| Hedgehog signaling in gastrointestinal development and disease., Harmon EB,Ko AH,Kim SK, Curr Mol Med. February 1, 2002; 2(1):1875-5666. |
|
Human disease-causing NOG missense mutations: effects on noggin secretion, dimer formation, and bone morphogenetic protein binding.,
Marcelino J,Sciortino CM,Romero MF,Ulatowski LM,Ballock RT,Economides AN,Eimon PM,Harland RM,Warman ML,
Proc Natl Acad Sci U S A. September 25, 2001; 98(20):1091-6490.
|
| Rapid functional analysis in Xenopus oocytes of Po protein adhesive interactions., Yoshida M,Colma DR, Neurochem Res. June 1, 2001; 26(6):1573-6903. |
| Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome., Ficker E,Thomas D,Viswanathan PC,Dennis AT,Priori SG,Napolitano C,Memmi M,Wible BA,Kaufman ES,Iyengar S,Schwartz PJ,Rudy Y,Brown AM, Am J Physiol Heart Circ Physiol. October 1, 2000; 279(4):0363-6135. |
| Signal transduction. Mating, channels and kidney cysts., Emmons SW,Somlo S, Nature. September 23, 1999; 401(6751):0143-5221. |
| Transcription repression by Xenopus ET and its human ortholog TBX3, a gene involved in ulnar-mammary syndrome., He Ml,Wen L,Campbell CE,Wu JY,Rao Y, Proc Natl Acad Sci U S A. August 31, 1999; 96(18):1091-6490. |
| Stilbenes and fenamates rescue the loss of I(KS) channel function induced by an LQT5 mutation and other IsK mutants., Abitbol I,Peretz A,Lerche C,Busch AE,Attali B, EMBO J. August 2, 1999; 18(15):0261-4189. |
| Point mutations in human GLI3 cause Greig syndrome., Wild A,Kalff-Suske M,Vortkamp A,Bornholdt D,König R,Grzeschik KH, Hum Mol Genet. October 1, 1997; 6(11):1460-2083. |
| The cloning of extracellular Ca(2+)-sensing receptors from parathyroid and kidney: molecular mechanisms of extracellular Ca(2+)-sensing., Brown EM,Pollak M,Chou YH,Seidman CE,Seidman JG,Hebert SC, J Nutr. July 1, 1995; 125(7 Suppl):1541-6100. |
|
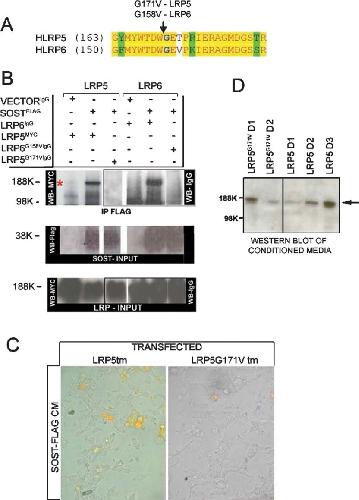
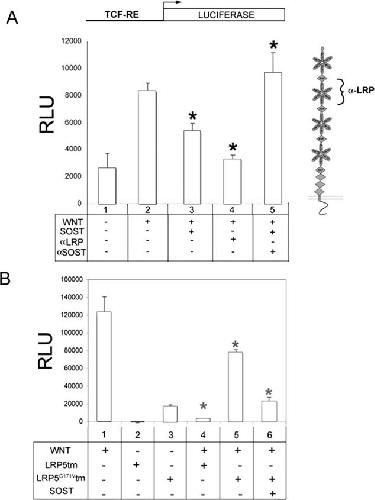
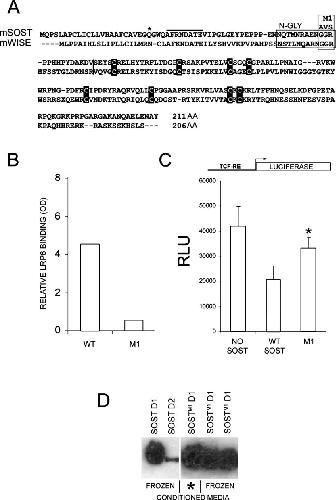
Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity.,
Ellies DL,Viviano B,McCarthy J,Rey JP,Itasaki N,Saunders S,Krumlauf R,
J Bone Miner Res. November 1, 2006; 21(11):0884-0431.
|
|
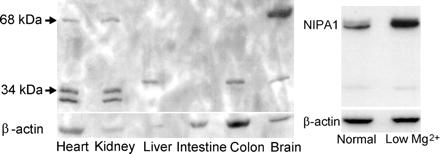
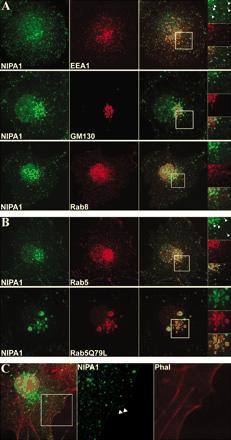
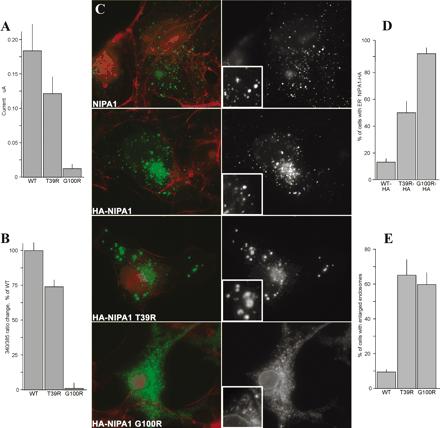
NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter.,
Goytain A,Hines RM,El-Husseini A,Quamme GA,
J Biol Chem. March 16, 2007; 282(11):1083-351X.
|
| FoxM1: at the crossroads of ageing and cancer., Laoukili J,Stahl M,Medema RH, Biochim Biophys Acta. January 1, 2007; 1775(1):0006-3002. |
| Mechanistic basis for the pathogenesis of long QT syndrome associated with a common splicing mutation in KCNQ1 gene., Tsuji K,Akao M,Ishii TM,Ohno S,Makiyama T,Takenaka K,Doi T,Haruna Y,Yoshida H,Nakashima T,Kita T,Horie M, J Mol Cell Cardiol. March 1, 2007; 42(3):1095-8584. |
|
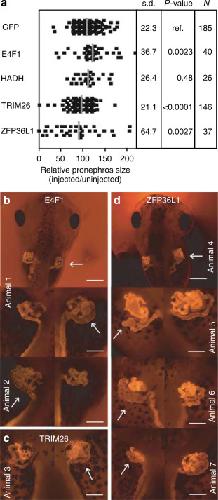
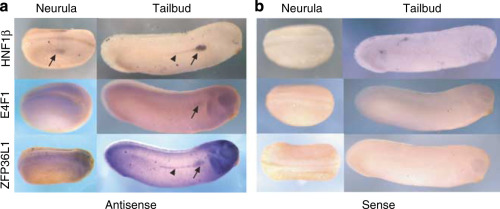
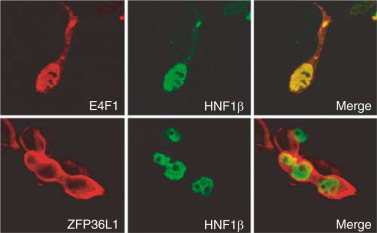
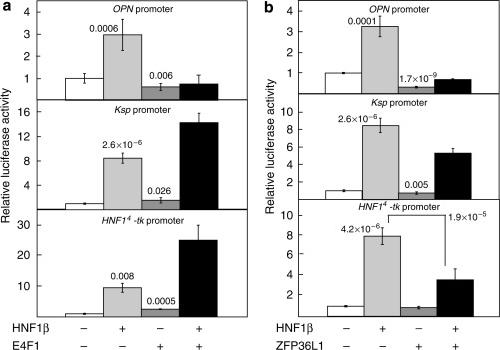
Transcription factor HNF1beta and novel partners affect nephrogenesis.,
Dudziak K,Mottalebi N,Senkel S,Edghill EL,Rosengarten S,Roose M,Bingham C,Ellard S,Ryffel GU,
Kidney Int. July 1, 2008; 74(2):1523-1755.
|
| Small molecule activator of the human epithelial sodium channel., Lu M,Echeverri F,Kalabat D,Laita B,Dahan DS,Smith RD,Xu H,Staszewski L,Yamamoto J,Ling J,Hwang N,Kimmich R,Li P,Patron E,Keung W,Patron A,Moyer BD, J Biol Chem. May 2, 2008; 283(18):1083-351X. |
| Biophysical properties of zebrafish ether-à-go-go related gene potassium channels., Scholz EP,Niemer N,Hassel D,Zitron E,Bürgers HF,Bloehs R,Seyler C,Scherer D,Thomas D,Kathöfer S,Katus HA,Rottbauer WA,Karle CA, Biochem Biophys Res Commun. April 3, 2009; 381(2):1090-2104. |
|
Vestibular asymmetry as the cause of idiopathic scoliosis: a possible answer from Xenopus.,
Lambert FM,Malinvaud D,Glaunès J,Bergot C,Straka H,Vidal PP,
J Neurosci. October 7, 2009; 29(40):1529-2401.
|
| Lessons from the lily pad: Using Xenopus to understand heart disease., Bartlett HL,Weeks DL, Drug Discov Today Dis Models. January 1, 2008; 5(3):1740-6757. |
|
A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels.,
Gustina AS,Trudeau MC,
Proc Natl Acad Sci U S A. August 4, 2009; 106(31):1091-6490.
|
|
EYA1 mutations associated with the branchio-oto-renal syndrome result in defective otic development in Xenopus laevis.,
Li Y,Manaligod JM,Weeks DL,
Biol Cell. February 17, 2010; 102(5):1768-322X.
|
| Polycystin-2 activity is controlled by transcriptional coactivator with PDZ binding motif and PALS1-associated tight junction protein., Duning K,Rosenbusch D,Schlüter MA,Tian Y,Kunzelmann K,Meyer N,Schulze U,Markoff A,Pavenstädt H,Weide T, J Biol Chem. October 29, 2010; 285(44):1083-351X. |
|
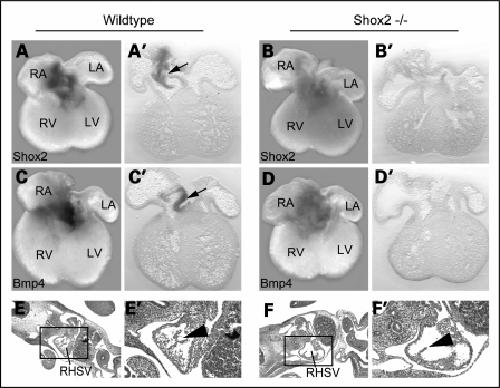
Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart.,
Puskaric S,Schmitteckert S,Mori AD,Glaser A,Schneider KU,Bruneau BG,Blaschke RJ,Steinbeisser H,Rappold G,
Hum Mol Genet. December 1, 2010; 19(23):1460-2083.
|
| Neurological disease mutations compromise a C-terminal ion pathway in the Na(+)/K(+)-ATPase., Poulsen H,Khandelia H,Morth JP,Bublitz M,Mouritsen OG,Egebjerg J,Nissen P, Nature. September 2, 2010; 467(7311):0143-5221. |
| Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α., Thorne CA,Hanson AJ,Schneider J,Tahinci E,Orton D,Cselenyi CS,Jernigan KK,Meyers KC,Hang BI,Waterson AG,Kim K,Melancon B,Ghidu VP,Sulikowski GA,LaFleur B,Salic A,Lee LA,Miller DM,Lee E,Lee E, Nat Chem Biol. November 1, 2010; 6(11):1552-4469. |
|
Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A.,
Ogawa Y,Nonaka Y,Goto T,Ohnishi E,Hiramatsu T,Kii I,Yoshida M,Ikura T,Onogi H,Shibuya H,Hosoya T,Ito N,Hagiwara M,
Nat Commun. October 5, 2010; 1:2041-1723.
|
|
Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria.,
Hernandez L,Roux KJ,Wong ES,Mounkes LC,Mutalif R,Navasankari R,Rai B,Cool S,Jeong JW,Wang H,Lee HS,Lee HS,Kozlov S,Grunert M,Keeble T,Jones CM,Meta MD,Young SG,Daar IO,Burke B,Perantoni AO,Stewart CL,
Dev Cell. September 14, 2010; 19(3):1878-1551.
|
|
Paraxial T-box genes, Tbx6 and Tbx1, are required for cranial chondrogenesis and myogenesis.,
Tazumi S,Yabe S,Uchiyama H,
Dev Biol. October 15, 2010; 346(2):1095-564X.
|
| Animal models for autosomal dominant frontal lobe epilepsy: on the origin of seizures., Steinlein OK, Expert Rev Neurother. December 1, 2010; 10(12):1744-8360. |
| Biophysical properties of mutant KCNQ1 S277L channels linked to hereditary long QT syndrome with phenotypic variability., Aidery P,Kisselbach J,Schweizer PA,Becker R,Katus HA,Thomas D, Biochim Biophys Acta. April 1, 2011; 1812(4):0006-3002. |
| Stomatin-deficient cryohydrocytosis results from mutations in SLC2A1: a novel form of GLUT1 deficiency syndrome., Flatt JF,Guizouarn H,Burton NM,Borgese F,Tomlinson RJ,Forsyth RJ,Baldwin SA,Levinson BE,Quittet P,Aguilar-Martinez P,Delaunay J,Stewart GW,Bruce LJ, Blood. November 10, 2011; 118(19):1528-0020. |
| Identification and functional characterization of KCNQ1 mutations around the exon 7-intron 7 junction affecting the splicing process., Tsuji-Wakisaka K,Akao M,Ishii TM,Ashihara T,Makiyama T,Ohno S,Toyoda F,Dochi K,Matsuura H,Horie M, Biochim Biophys Acta. November 1, 2011; 1812(11):0006-3002. |
| Structural basis of slow activation gating in the cardiac I Ks channel complex., Strutz-Seebohm N,Pusch M,Wolf S,Stoll R,Tapken D,Gerwert K,Attali B,Seebohm G, Cell Physiol Biochem. January 1, 2011; 27(5):1421-9778. |
|
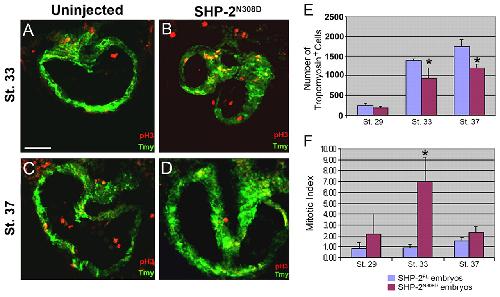
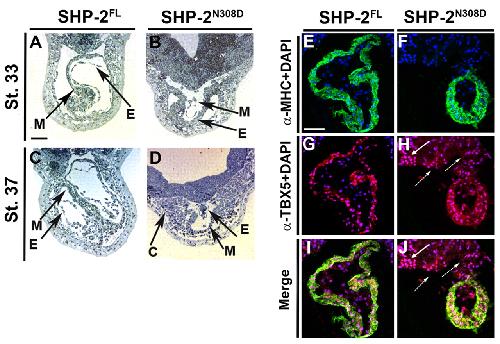
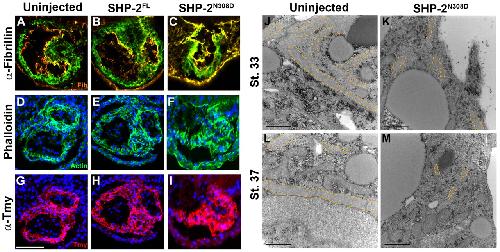
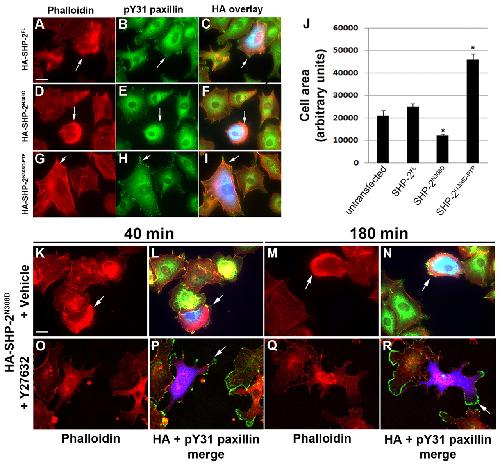
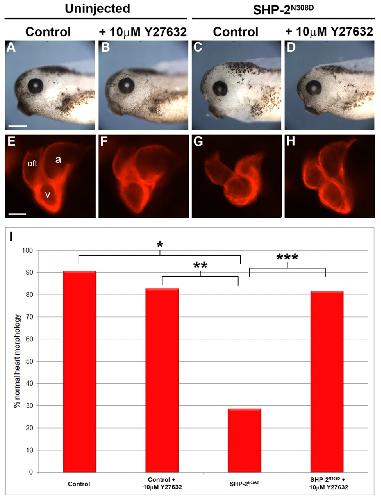
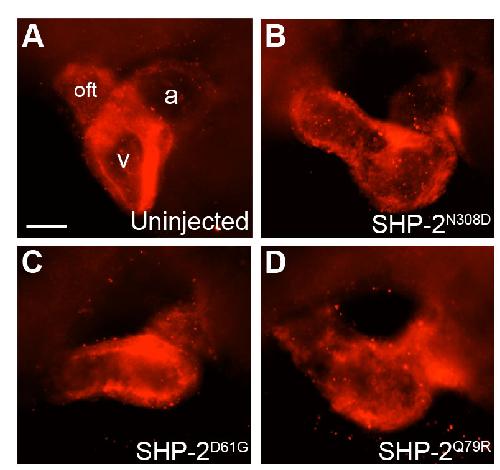
SHP-2 acts via ROCK to regulate the cardiac actin cytoskeleton.,
Langdon Y,Tandon P,Paden E,Duddy J,Taylor JM,Conlon FL,
Development. March 1, 2012; 139(5):1477-9129.
|
|
Williams Syndrome Transcription Factor is critical for neural crest cell function in Xenopus laevis.,
Barnett C,Yazgan O,Kuo HC,Malakar S,Thomas T,Fitzgerald A,Harbour W,Henry JJ,Krebs JE,
Mech Dev. January 1, 2012; 129(9-12):1872-6356.
|
|
Impaired ion channel function related to a common KCNQ1 mutation - implications for risk stratification in long QT syndrome 1.,
Aidery P,Kisselbach J,Schweizer PA,Becker R,Katus HA,Thomas D,
Gene. December 10, 2012; 511(1):1879-0038.
|
| Amphibian oocyte nuclei expressing lamin A with the progeria mutation E145K exhibit an increased elastic modulus., Kaufmann A,Heinemann F,Radmacher M,Stick R, Nucleus. January 1, 2011; 2(4):1949-1042. |
| Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation., Streets AJ,Wessely O,Peters DJ,Ong AC, Hum Mol Genet. May 15, 2013; 22(10):1460-2083. |
|
Connexins in epidermal homeostasis and skin disease.,
Scott CA,Tattersall D,O'Toole EA,Kelsell DP,
Biochim Biophys Acta. August 1, 2012; 1818(8):0006-3002.
|
|
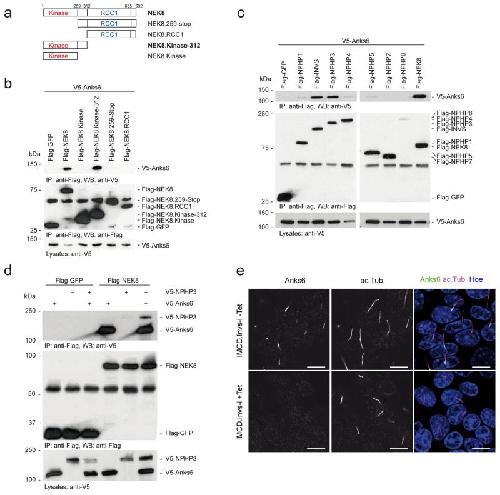
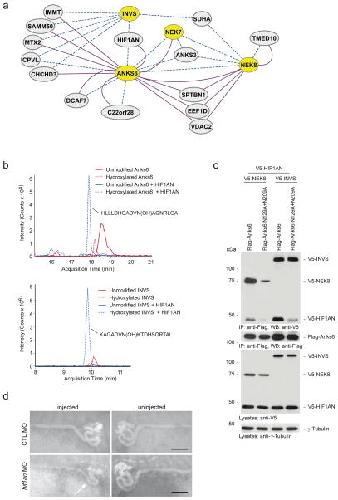
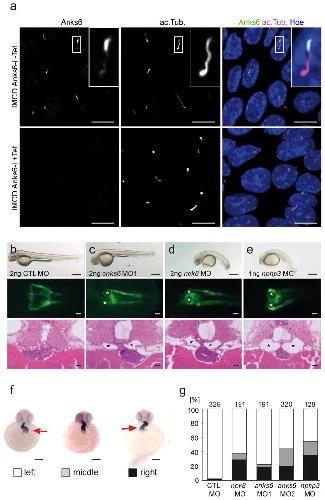
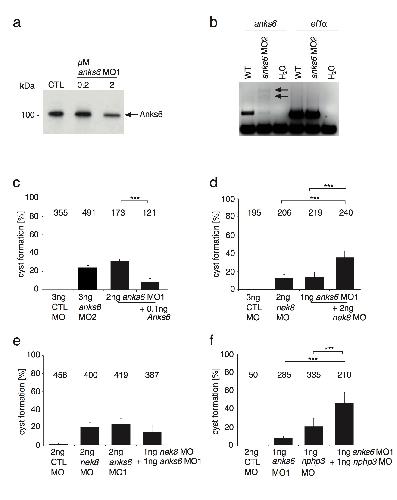
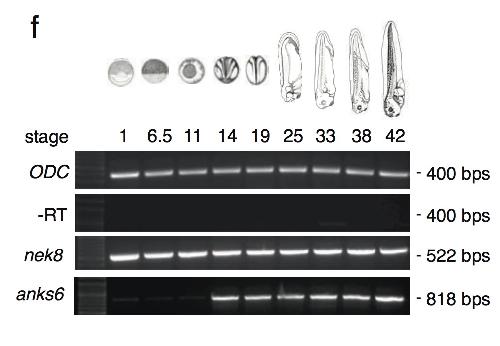
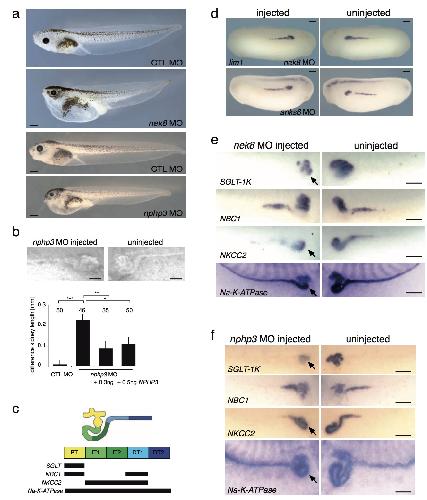
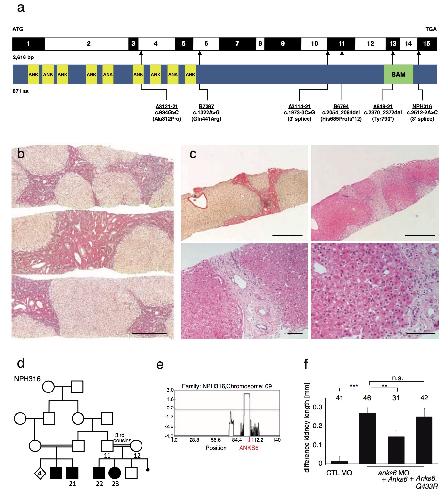
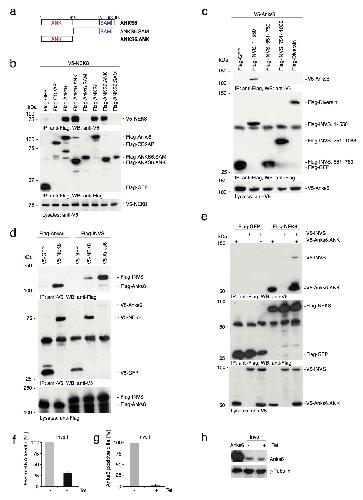
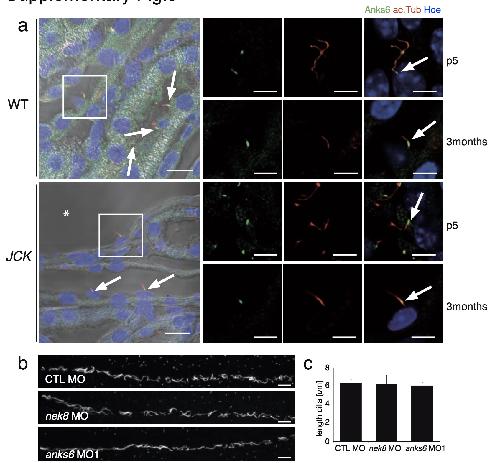
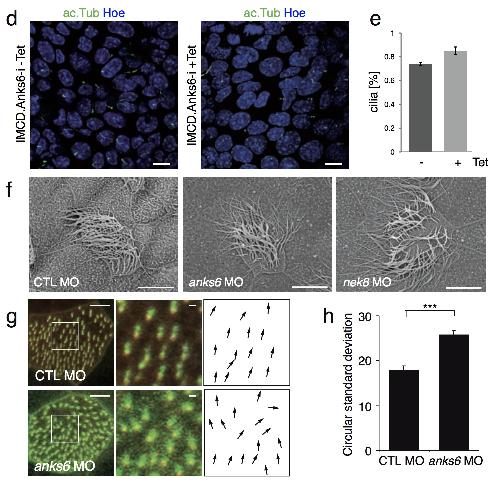
ANKS6 is a central component of a nephronophthisis module linking NEK8 to INVS and NPHP3.,
Hoff S,Halbritter J,Epting D,Frank V,Nguyen TM,van Reeuwijk J,Boehlke C,Schell C,Yasunaga T,Helmstädter M,Mergen M,Filhol E,Boldt K,Horn N,Ueffing M,Otto EA,Eisenberger T,Elting MW,van Wijk JA,Bockenhauer D,Sebire NJ,Rittig S,Vyberg M,Ring T,Pohl M,Pape L,Neuhaus TJ,Elshakhs NA,Koon SJ,Harris PC,Grahammer F,Huber TB,Kuehn EW,Kramer-Zucker A,Bolz HJ,Roepman R,Saunier S,Walz G,Hildebrandt F,Bergmann C,Lienkamp SS,
Nat Genet. August 1, 2013; 45(8):1546-1718.
|
|
The variant hERG/R148W associated with LQTS is a mutation that reduces current density on co-expression with the WT.,
Mechakra A,Vincent Y,Chevalier P,Millat G,Ficker E,Jastrzebski M,Poulin H,Pouliot V,Chahine M,Christé G,
Gene. February 25, 2014; 536(2):1879-0038.
|
|
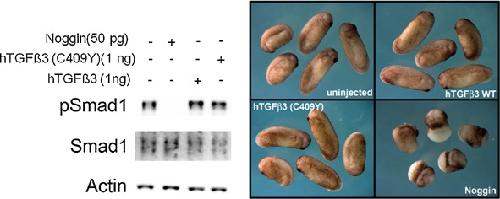
A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with Marfan and Loeys-Dietz syndrome.,
Rienhoff HY,Yeo CY,Morissette R,Khrebtukova I,Melnick J,Luo S,Leng N,Kim YJ,Schroth G,Westwick J,Vogel H,McDonnell N,Hall JG,Whitman M,
Am J Med Genet A. August 1, 2013; 161A(8):1552-4833.
|
|
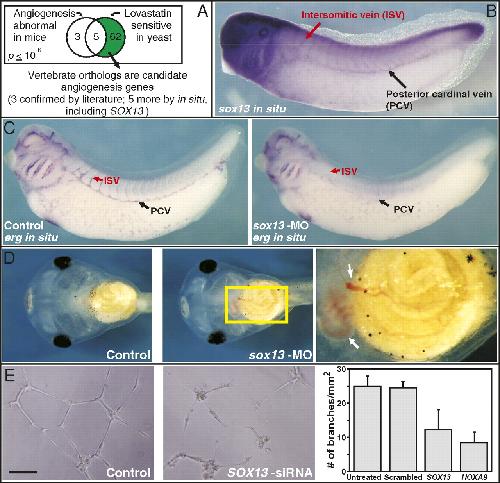
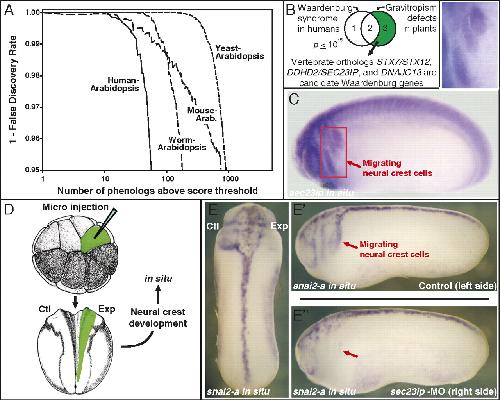
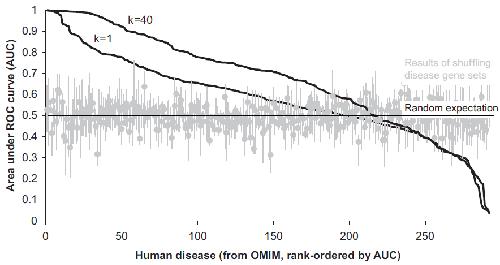
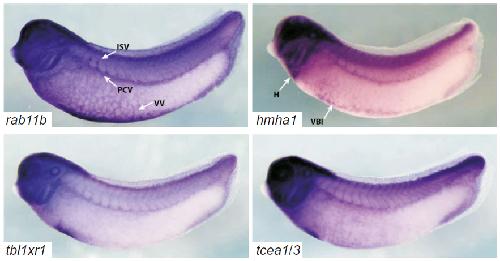
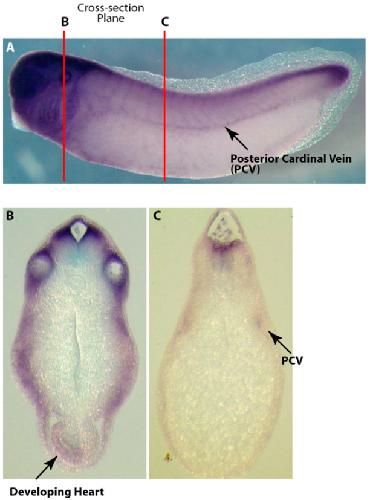
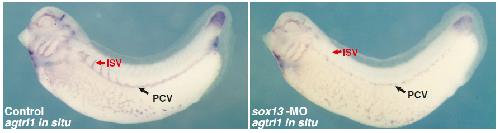
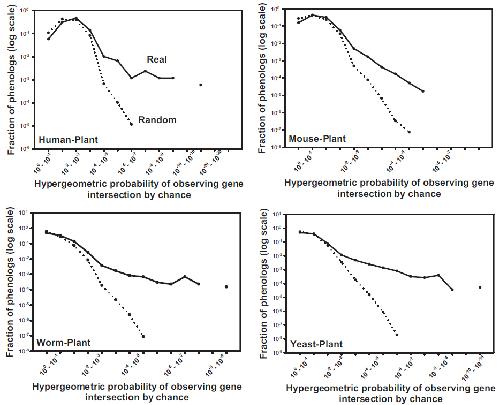
Systematic discovery of nonobvious human disease models through orthologous phenotypes.,
McGary KL,Park TJ,Woods JO,Cha HJ,Wallingford JB,Marcotte EM,
Proc Natl Acad Sci U S A. April 6, 2010; 107(14):1091-6490.
|
|
Aberrant connexin26 hemichannels underlying keratitis-ichthyosis-deafness syndrome are potently inhibited by mefloquine.,
Levit NA,Sellitto C,Wang HZ,Li L,Srinivas M,Brink PR,White TW,
J Invest Dermatol. April 1, 2015; 135(4):1523-1747.
|
|
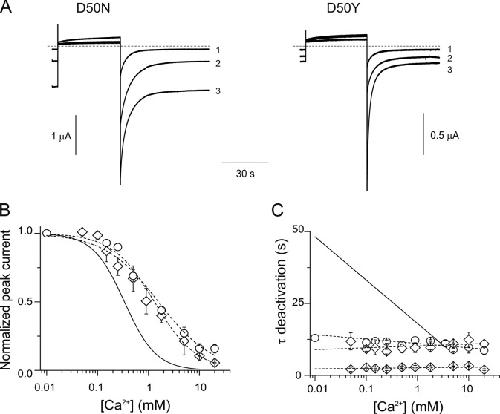
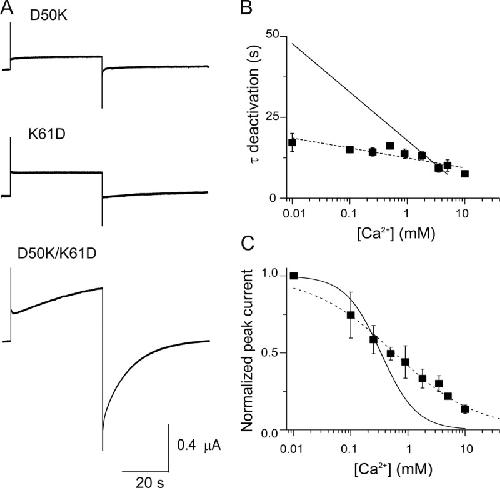
Insights on the mechanisms of Ca(2+) regulation of connexin26 hemichannels revealed by human pathogenic mutations (D50N/Y).,
Lopez W,Gonzalez J,Liu Y,Harris AL,Contreras JE,
J Gen Physiol. July 1, 2013; 142(1):1540-7748.
|
|
High incidence of functional ion-channel abnormalities in a consecutive Long QT cohort with novel missense genetic variants of unknown significance.,
Steffensen AB,Refaat MM,David JP,Mujezinovic A,Calloe K,Wojciak J,Nussbaum RL,Scheinman MM,Schmitt N,
Sci Rep. January 12, 2015; 5:2045-2322.
|
| The UNC-45 myosin chaperone: from worms to flies to vertebrates., Lee CF,Lee CF,Lee CF,Melkani GC,Bernstein SI, Int Rev Cell Mol Biol. January 1, 2014; 313:1937-6448. |
|
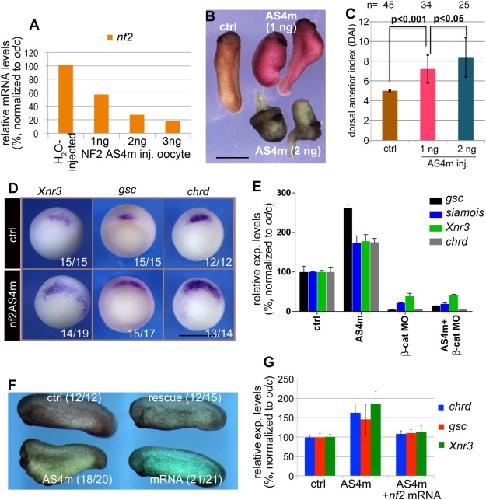
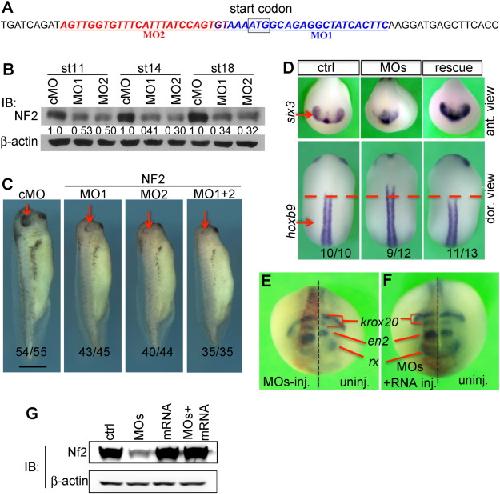
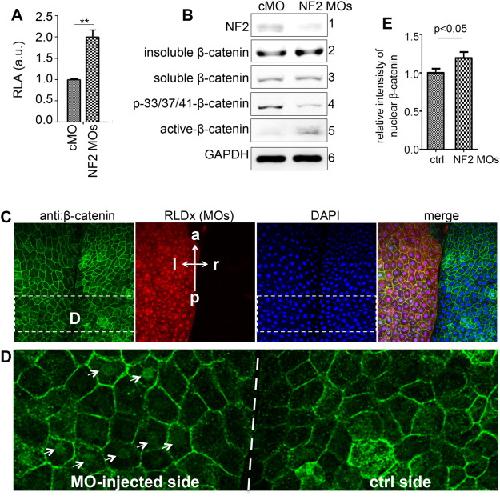
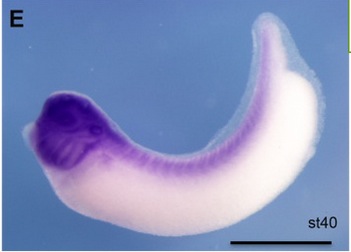
NF2/Merlin is required for the axial pattern formation in the Xenopus laevis embryo.,
Zhu X,Min Z,Tan R,Tao Q,Tao Q,
Mech Dev. November 1, 2015; 138 Pt 3:1872-6356.
|
|
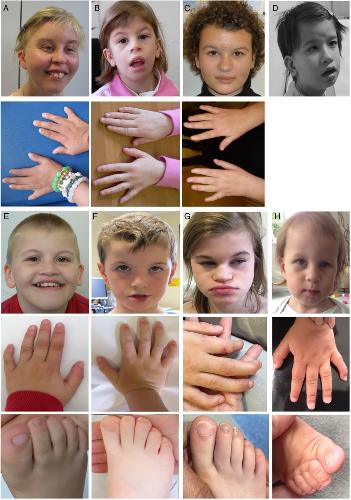
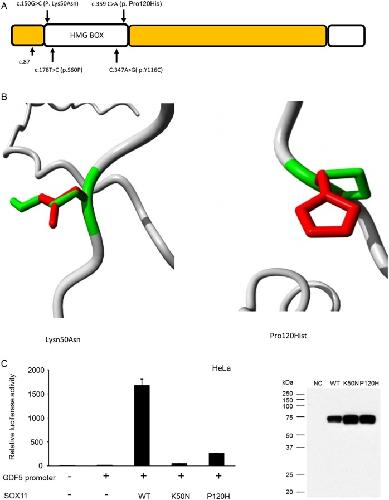
Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome.,
Hempel A,Pagnamenta AT,Blyth M,Mansour S,McConnell V,Kou I,Ikegawa S,Tsurusaki Y,Matsumoto N,Lo-Castro A,Plessis G,Albrecht B,Battaglia A,Taylor JC,Howard MF,Keays D,Sohal AS,Kühl SJ,Kühl SJ,Kini U,McNeill A,
J Med Genet. March 1, 2016; 53(3):1468-6244.
|
| Distinctive effects of nicotinic receptor intracellular-loop mutations associated with nocturnal frontal lobe epilepsy., Weltzin MM,Lindstrom JM,Lukas RJ,Whiteaker P, Neuropharmacology. March 1, 2016; 102:1873-7064. |
|
Xenopus as a model system for studying pancreatic development and diabetes.,
Kofent J,Spagnoli FM,
Semin Cell Dev Biol. March 1, 2016; 51:1096-3634.
|
|
Using Xenopus to study genetic kidney diseases.,
Lienkamp SS,
Semin Cell Dev Biol. March 1, 2016; 51:1096-3634.
|
| Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions., Ruffolo G,Iyer A,Cifelli P,Roseti C,Mühlebner A,van Scheppingen J,Scholl T,Hainfellner JA,Feucht M,Krsek P,Zamecnik J,Jansen FE,Spliet WG,Limatola C,Aronica E,Palma E, Neurobiol Dis. November 1, 2016; 95:1095-953X. |
|
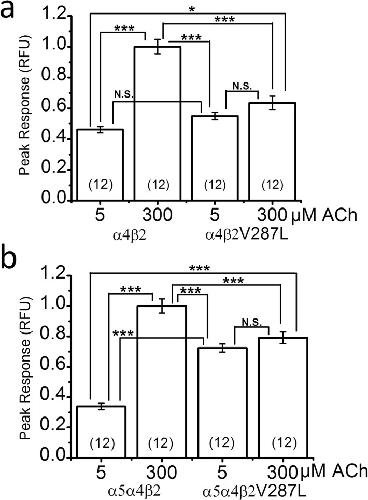
Mutation Linked to Autosomal Dominant Nocturnal Frontal Lobe Epilepsy Reduces Low-Sensitivity α4β2, and Increases α5α4β2, Nicotinic Receptor Surface Expression.,
Nichols WA,Henderson BJ,Marotta CB,Yu CY,Richards C,Dougherty DA,Lester HA,Cohen BN,
PLoS One. January 1, 2016; 11(6):1932-6203.
|
|
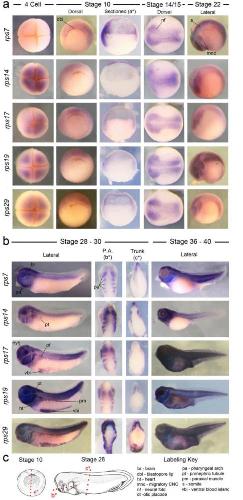
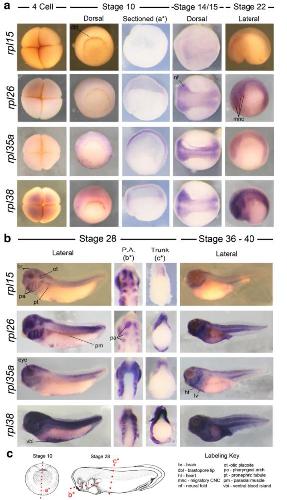
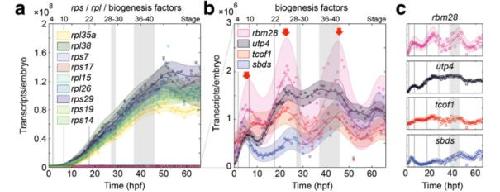
Expression of ribosomopathy genes during Xenopus tropicalis embryogenesis.,
Robson A,Owens ND,Baserga SJ,Khokha MK,Griffin JN,
BMC Dev Biol. October 26, 2016; 16(1):1471-213X.
|
|
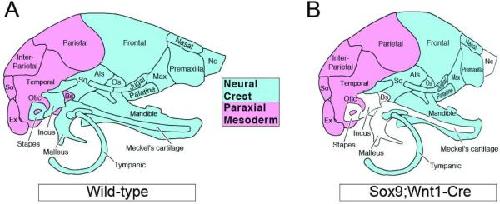
Sox9 function in craniofacial development and disease.,
Lee YH,Lee YH,Saint-Jeannet JP,
Genesis. April 1, 2011; 49(4):1526-968X.
|
| Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome)., Tristani-Firouzi M,Jensen JL,Donaldson MR,Sansone V,Meola G,Hahn A,Bendahhou S,Kwiecinski H,Fidzianska A,Plaster N,Fu YH,Ptacek LJ,Tawil R, J Clin Invest. August 1, 2002; 110(3):1558-8238. |
|
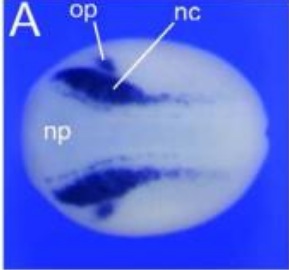
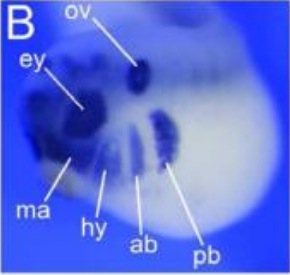
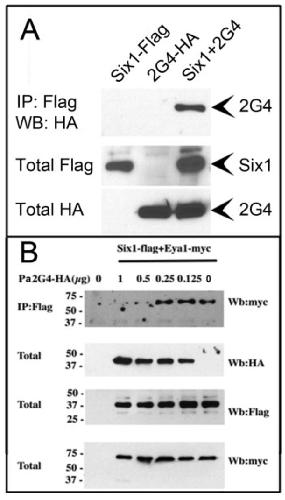
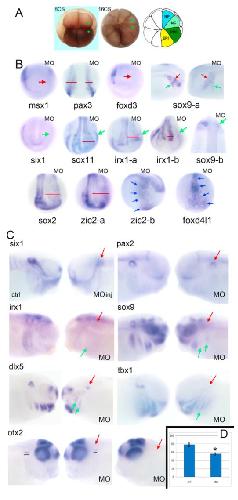
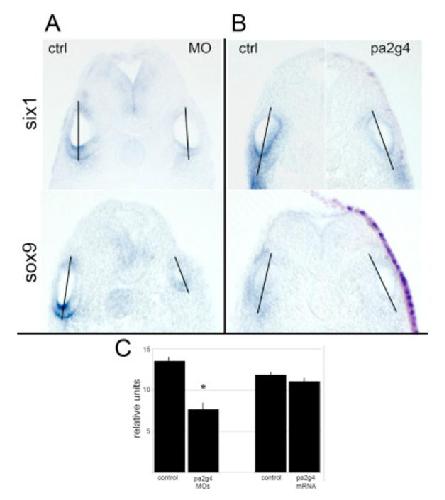
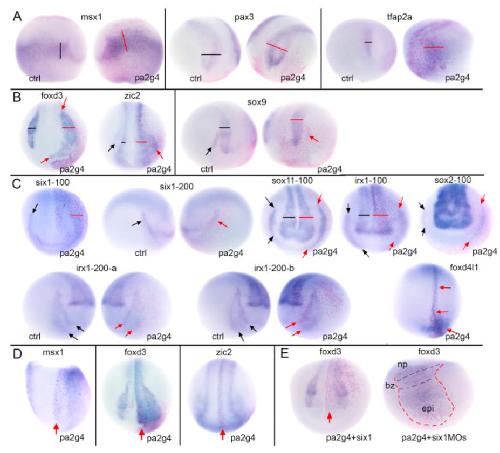
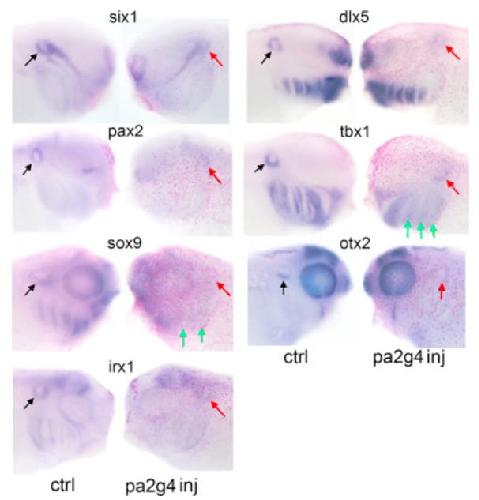
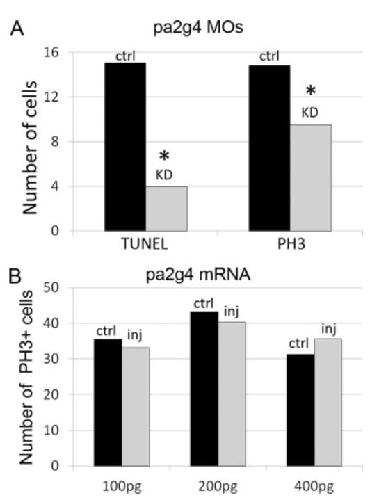
Pa2G4 is a novel Six1 co-factor that is required for neural crest and otic development.,
Neilson KM,Abbruzzesse G,Kenyon K,Bartolo V,Krohn P,Alfandari D,Alfandari D,Moody SA,
Dev Biol. January 15, 2017; 421(2):1095-564X.
|
| Heterozygous Pathogenic Variant in DACT1 Causes an Autosomal-Dominant Syndrome with Features Overlapping Townes-Brocks Syndrome., Webb BD,Metikala S,Wheeler PG,Sherpa MD,Houten SM,Horb ME,Schadt EE, Hum Mutat. April 1, 2017; 38(4):1098-1004. |
| Mutations in GABRB3: From febrile seizures to epileptic encephalopathies., Møller RS,Wuttke TV,Helbig I,Marini C,Johannesen KM,Brilstra EH,Vaher U,Borggraefe I,Talvik I,Talvik T,Kluger G,Francois LL,Lesca G,de Bellescize J,Blichfeldt S,Chatron N,Holert N,Jacobs J,Swinkels M,Betzler C,Syrbe S,Nikanorova M,Myers CT,Larsen LH,Vejzovic S,Pendziwiat M,von Spiczak S,Hopkins S,Dubbs H,Mang Y,Mukhin K,Holthausen H,van Gassen KL,Dahl HA,Tommerup N,Mefford HC,Rubboli G,Guerrini R,Lemke JR,Lerche H,Muhle H,Maljevic S, Neurology. January 31, 2017; 88(5):1526-632X. |
|
TALENs and CRISPR/Cas9 fuel genetically engineered clinically relevant Xenopus tropicalis tumor models.,
Naert T,Van Nieuwenhuysen T,Vleminckx K,Vleminckx K,
Genesis. January 1, 2017; 55(1-2):1526-968X.
|
| Modeling human craniofacial disorders in Xenopus., Dubey A,Saint-Jeannet JP, Curr Pathobiol Rep. March 1, 2017; 5(1):2167-485X. |
|
Mechanosensory Stimulation Evokes Acute Concussion-Like Behavior by Activating GIRKs Coupled to Muscarinic Receptors in a Simple Vertebrate.,
Li WC,Zhu XY,Ritson E,
eNeuro. January 1, 2017; 4(2):2373-2822.
|
| The Sorting Nexin 3 Retromer Pathway Regulates the Cell Surface Localization and Activity of a Wnt-Activated Polycystin Channel Complex., Feng S,Streets AJ,Nesin V,Tran U,Nie H,Onopiuk M,Wessely O,Tsiokas L,Ong ACM, J Am Soc Nephrol. October 1, 2017; 28(10):1533-3450. |
|
Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy.,
McTague A,Nair U,Malhotra S,Meyer E,Trump N,Gazina EV,Papandreou A,Ngoh A,Ackermann S,Ambegaonkar G,Appleton R,Desurkar A,Eltze C,Kneen R,Kumar AV,Lascelles K,Montgomery T,Ramesh V,Samanta R,Scott RH,Tan J,Whitehouse W,Poduri A,Scheffer IE,Chong WKK,Cross JH,Topf M,Petrou S,Kurian MA,
Neurology. January 2, 2018; 90(1):1526-632X.
|
|
Polycystin 1 loss of function is directly linked to an imbalance in G-protein signaling in the kidney.,
Zhang B,Tran U,Wessely O,
Development. March 22, 2018; 145(6):1477-9129.
|
|
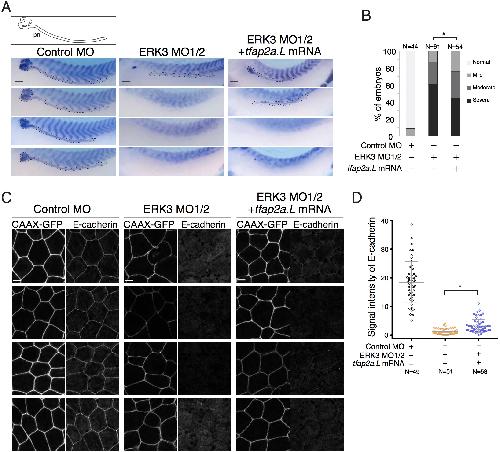
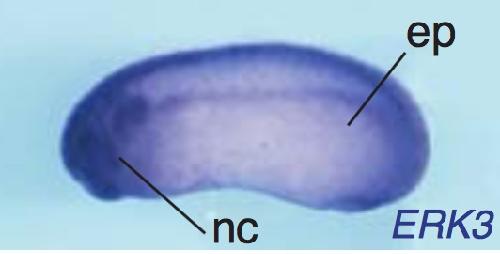
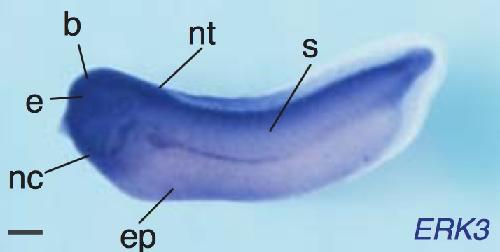
The atypical mitogen-activated protein kinase ERK3 is essential for establishment of epithelial architecture.,
Takahashi C,Miyatake K,Kusakabe M,Nishida E,
J Biol Chem. June 1, 2018; 293(22):1083-351X.
|
| Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders., Calo E,Gu B,Bowen ME,Aryan F,Zalc A,Liang J,Flynn RA,Swigut T,Chang HY,Attardi LD,Wysocka J, Nature. February 1, 2018; 554(7690):0143-5221. |
| A novel GABAergic dysfunction in human Dravet syndrome., Ruffolo G,Cifelli P,Roseti C,Thom M,van Vliet EA,Limatola C,Aronica E,Palma E, Epilepsia. November 1, 2018; 59(11):1528-1167. |
|
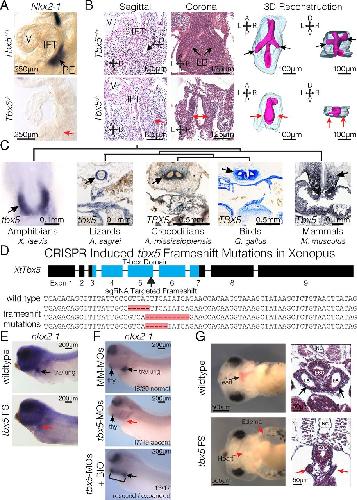
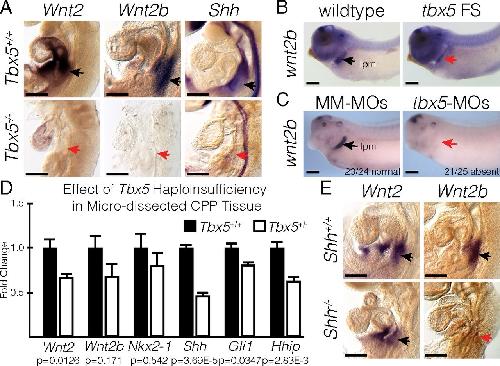
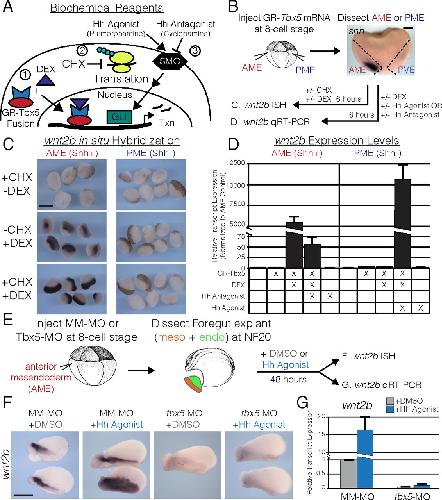
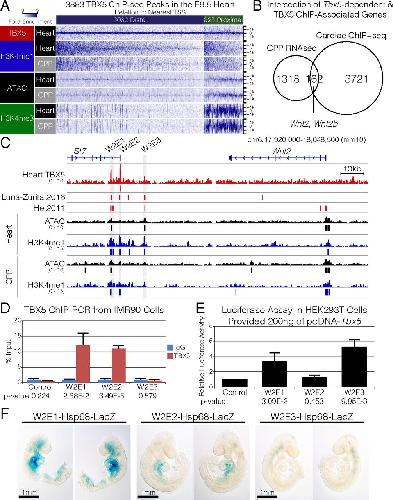
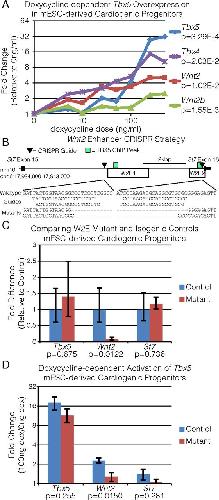
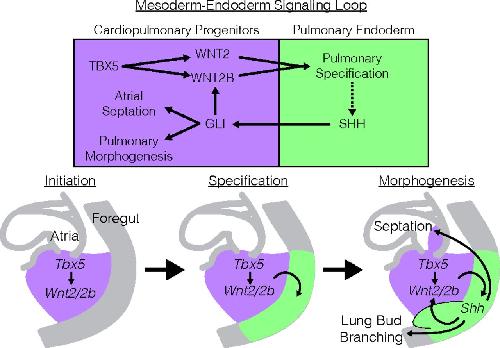
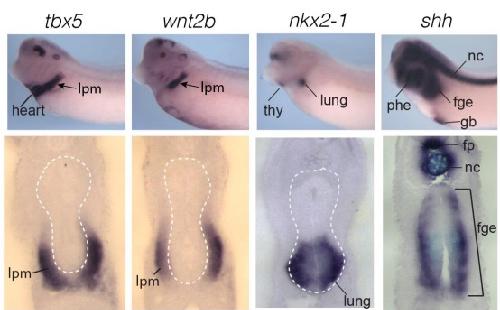
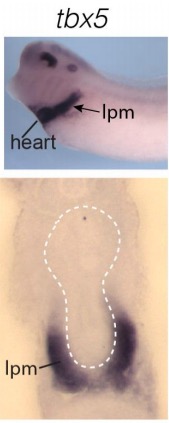
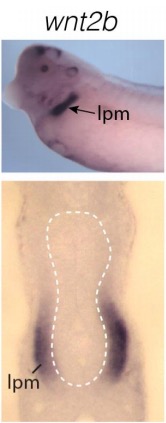
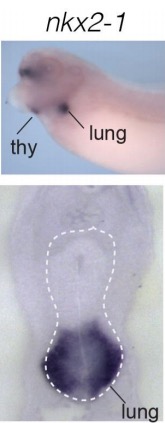
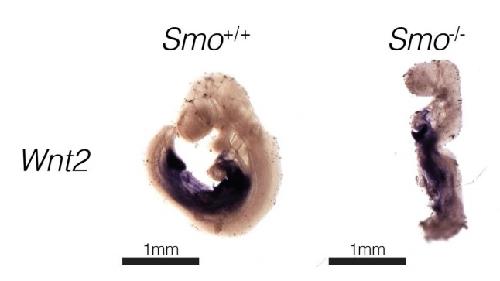
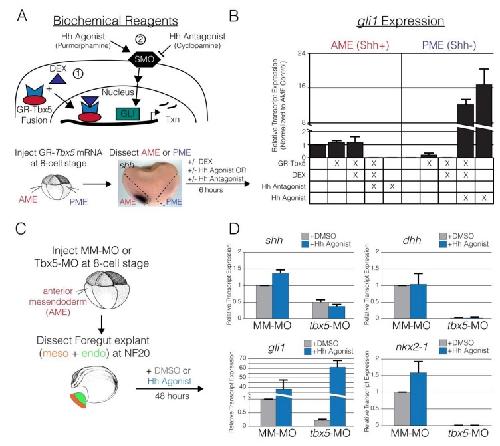
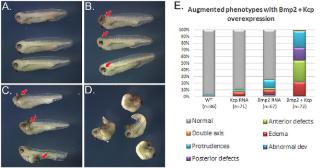
Evolutionarily conserved Tbx5-Wnt2/2b pathway orchestrates cardiopulmonary development.,
Steimle JD,Rankin SA,Rankin SA,Slagle CE,Bekeny J,Rydeen AB,Chan SS,Kweon J,Yang XH,Ikegami K,Nadadur RD,Rowton M,Hoffmann AD,Lazarevic S,Thomas W,Boyle Anderson EAT,Horb ME,Luna-Zurita L,Ho RK,Kyba M,Jensen B,Zorn AM,Conlon FL,Moskowitz IP,
Proc Natl Acad Sci U S A. November 6, 2018; 115(45):1091-6490.
|
| A novel autosomal recessive GJB2-associated disorder: Ichthyosis follicularis, bilateral severe sensorineural hearing loss, and punctate palmoplantar keratoderma., Youssefian L,Vahidnezhad H,Saeidian AH,Mahmoudi H,Karamzadeh R,Kariminejad A,Huang J,Li L,Jannace TF,Fortina P,Zeinali S,White TW,Uitto J, Hum Mutat. February 1, 2019; 40(2):1098-1004. |
|
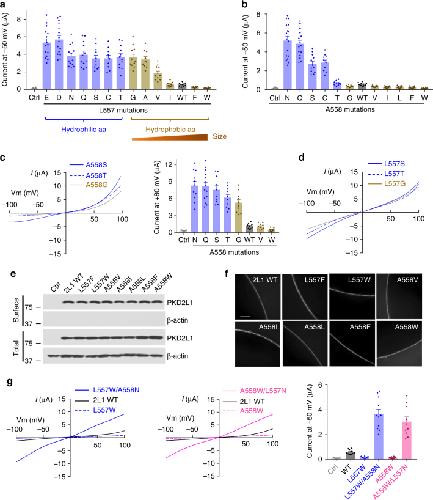
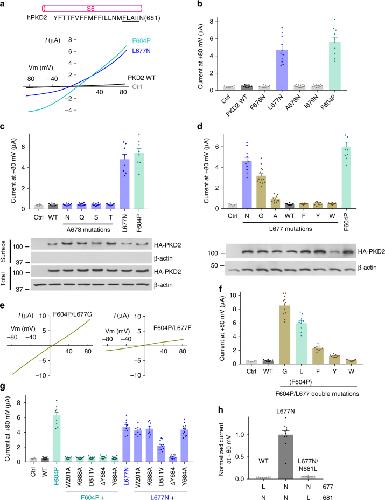
Hydrophobic pore gates regulate ion permeation in polycystic kidney disease 2 and 2L1 channels.,
Zheng W,Yang X,Hu R,Cai R,Hofmann L,Wang Z,Hu Q,Liu X,Bulkley D,Yu Y,Tang J,Flockerzi V,Cao Y,Cao Y,Cao E,Chen XZ,
Nat Commun. June 13, 2018; 9(1):2041-1723.
|
| De Novo SOX4 Variants Cause a Neurodevelopmental Disease Associated with Mild Dysmorphism., Zawerton A,Yao B,Yeager JP,Pippucci T,Haseeb A,Smith JD,Wischmann L,Kühl SJ,Dean JCS,Pilz DT,Holder SE,McNeill A,Graziano C,Lefebvre V, Am J Hum Genet. February 7, 2019; 104(2):1537-6605. |
|
Modeling congenital kidney diseases in Xenopus laevis.,
Blackburn ATM,Miller RK,
Dis Model Mech. April 9, 2019; 12(4):1754-8411.
|
|
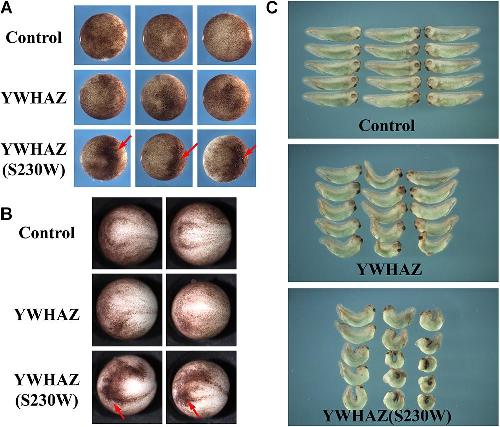
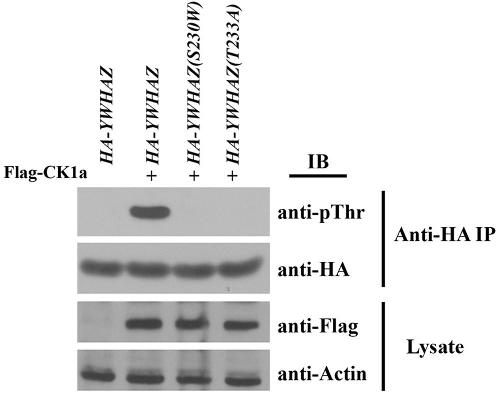
A YWHAZ Variant Associated With Cardiofaciocutaneous Syndrome Activates the RAF-ERK Pathway.,
Popov IK,Hiatt SM,Whalen S,Keren B,Ruivenkamp C,van Haeringen A,Chen MJ,Cooper GM,Korf BR,Chang C,
Front Physiol. January 1, 2019; 10:1664-042X.
|
|
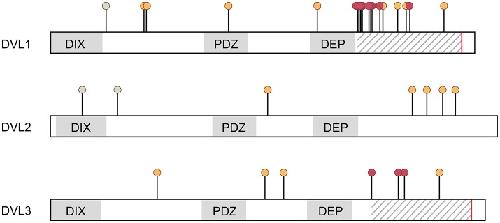
Dishevelled Paralogs in Vertebrate Development: Redundant or Distinct?,
Gentzel M,Schambony A,
Front Cell Dev Biol. May 26, 2017; 5:2296-634X.
|
|
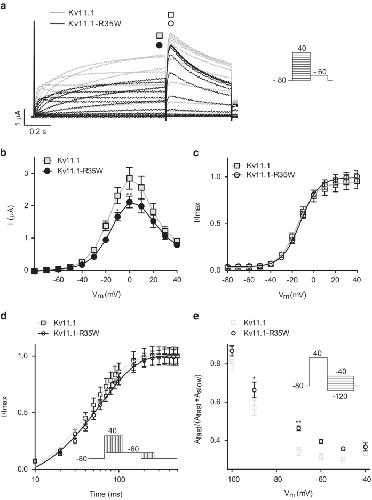
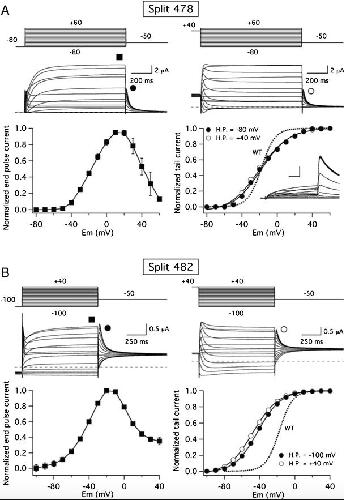
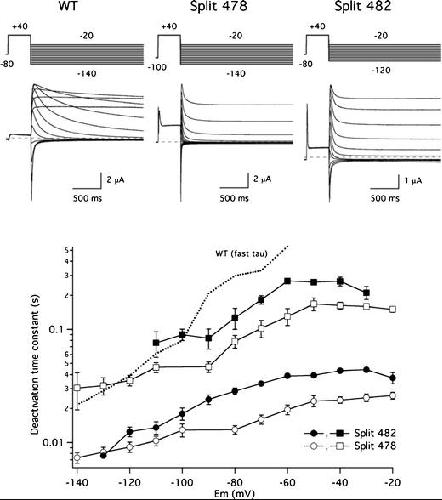
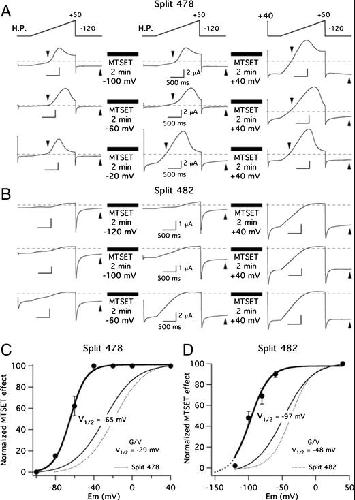
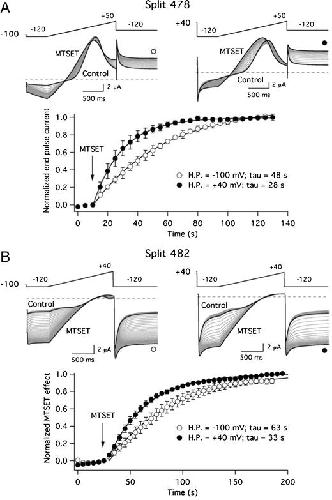
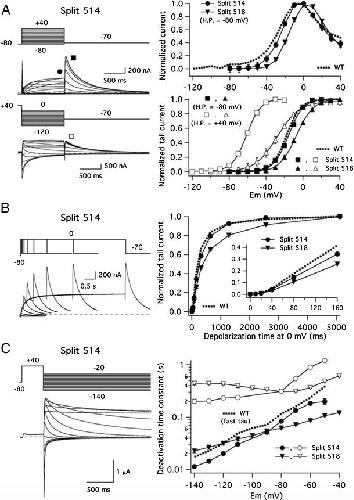
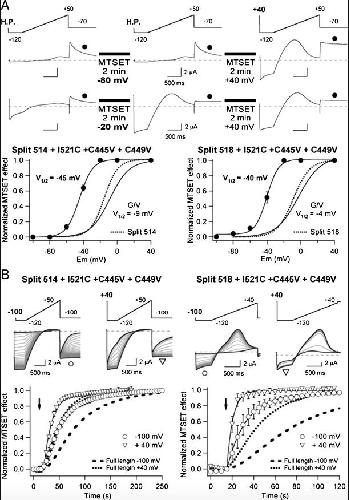
Functional characterization of Kv11.1 (hERG) potassium channels split in the voltage-sensing domain.,
de la Peña P,Domínguez P,Barros F,
Pflugers Arch. July 1, 2018; 470(7):1432-2013.
|
|
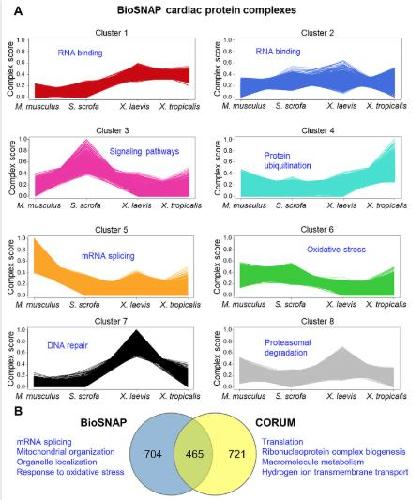
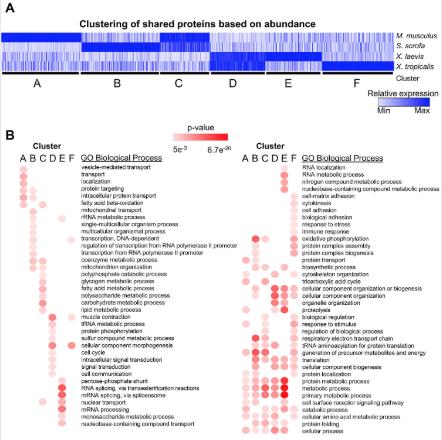
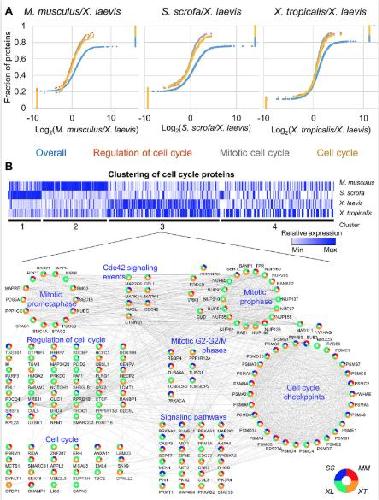
Conservation and divergence of protein pathways in the vertebrate heart.,
Federspiel JD,Tandon P,Wilczewski CM,Wasson L,Herring LE,Venkatesh SS,Cristea IM,Conlon FL,
PLoS Biol. September 6, 2019; 17(9):1545-7885.
|
|
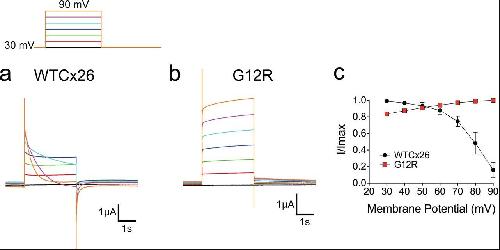
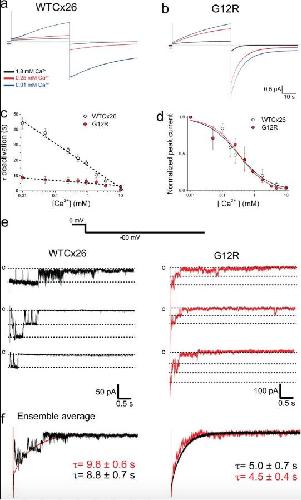
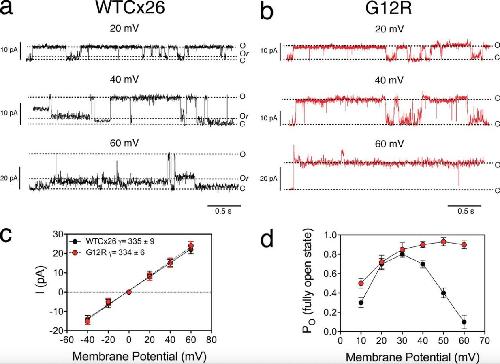
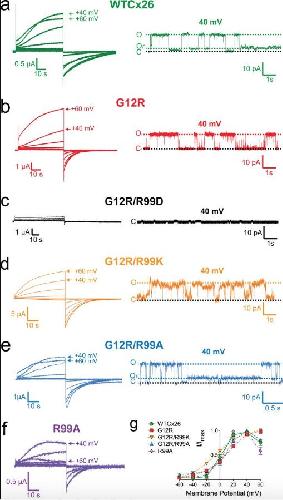
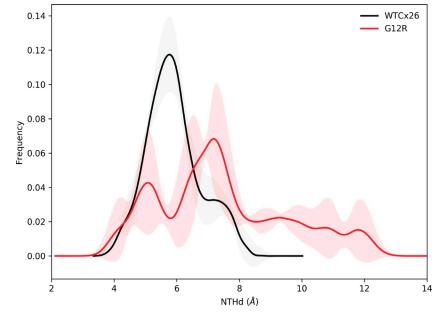
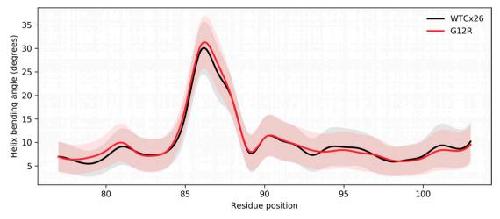
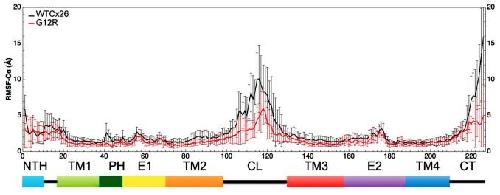
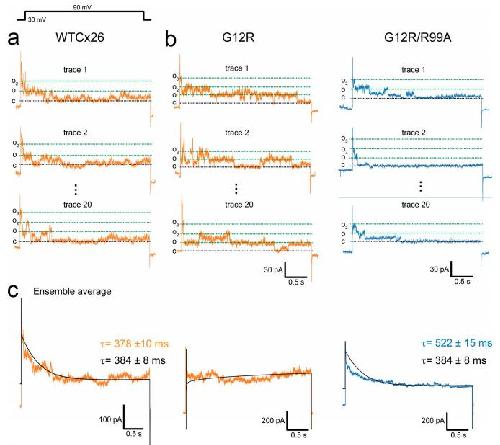
The syndromic deafness mutation G12R impairs fast and slow gating in Cx26 hemichannels.,
García IE,Villanelo F,Contreras GF,Pupo A,Pinto BI,Contreras JE,Pérez-Acle T,Alvarez O,Latorre R,Martínez AD,González C,
J Gen Physiol. May 7, 2018; 150(5):1540-7748.
|
|
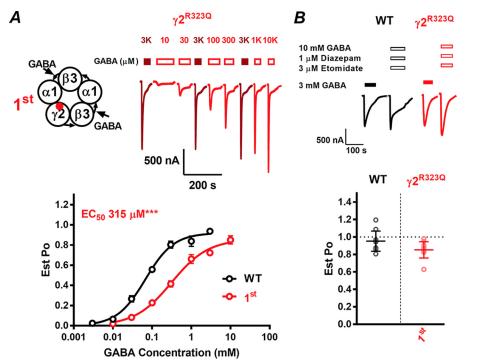
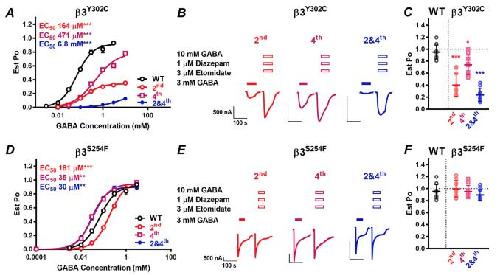
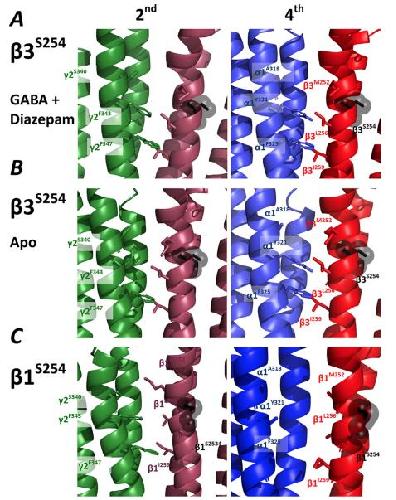
Functional genomics of epilepsy-associated mutations in the GABAA receptor subunits reveal that one mutation impairs function and two are catastrophic.,
Absalom NL,Ahring PK,Liao VW,Balle T,Jiang T,Anderson LL,Arnold JC,McGregor IS,Bowen MT,Chebib M,
J Biol Chem. April 12, 2019; 294(15):1083-351X.
|
|
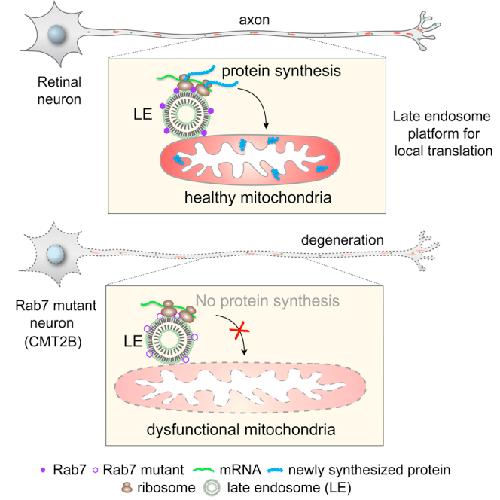
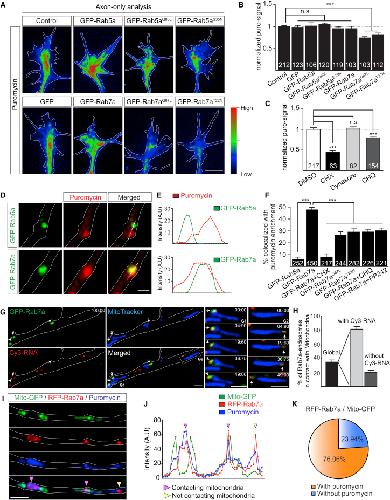
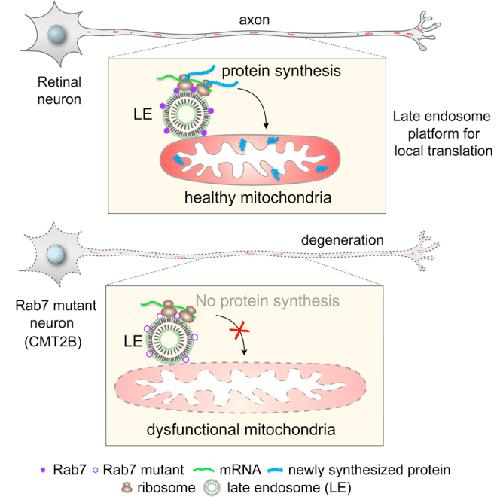
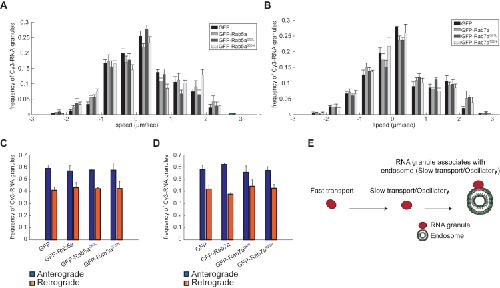
Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons.,
Cioni JM,Lin JQ,Holtermann AV,Koppers M,Jakobs MAH,Azizi A,Turner-Bridger B,Shigeoka T,Franze K,Harris WA,Holt CE,
Cell. January 10, 2019; 176(1-2):1097-4172.
|
|
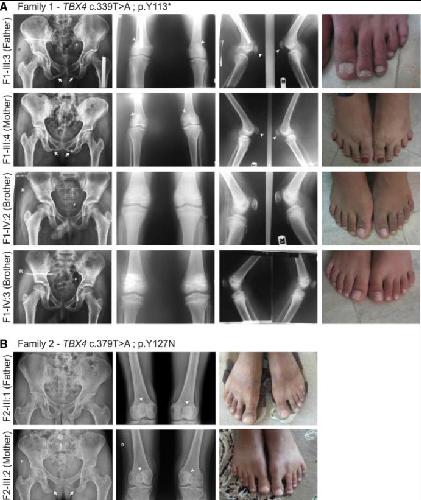
Homozygous Null TBX4 Mutations Lead to Posterior Amelia with Pelvic and Pulmonary Hypoplasia.,
Kariminejad A,Szenker-Ravi E,Lekszas C,Tajsharghi H,Moslemi AR,Naert T,Tran HT,Ahangari F,Rajaei M,Nasseri M,Haaf T,Azad A,Superti-Furga A,Maroofian R,Ghaderi-Sohi S,Najmabadi H,Abbaszadegan MR,Vleminckx K,Vleminckx K,Nikuei P,Reversade B,
Am J Hum Genet. December 5, 2019; 105(6):1537-6605.
|
|
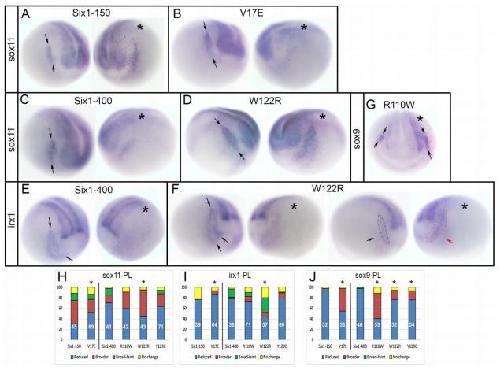
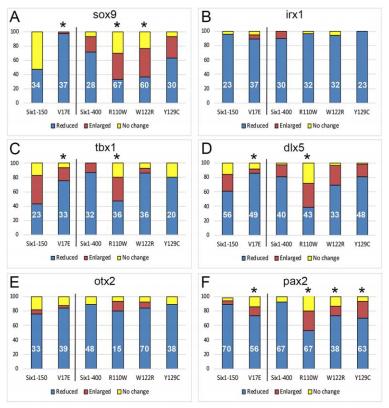
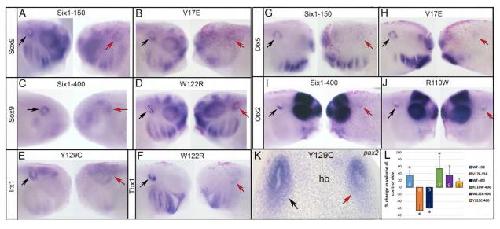
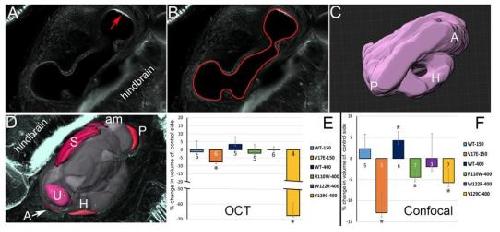
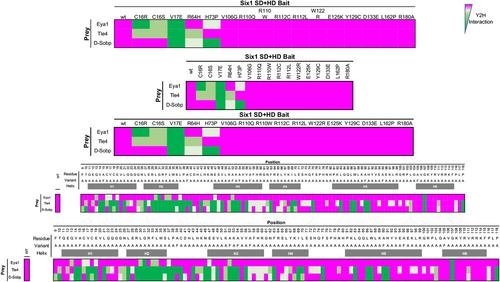
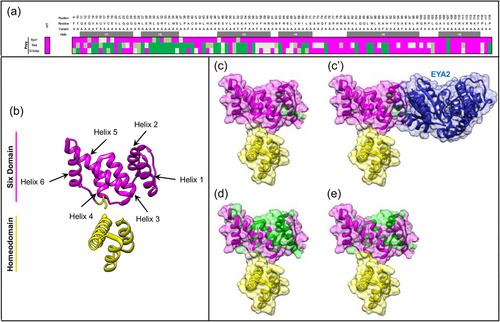
Six1 proteins with human branchio-oto-renal mutations differentially affect cranial gene expression and otic development.,
Shah AM,Krohn P,Baxi AB,Tavares ALP,Sullivan CH,Chillakuru YR,Majumdar HD,Neilson KM,Moody SA,
Dis Model Mech. March 3, 2020; 13(3):1754-8411.
|
|
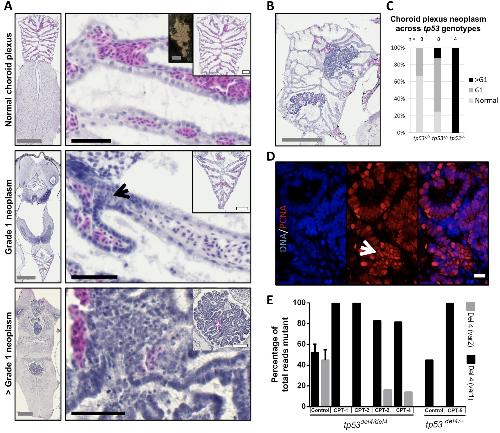
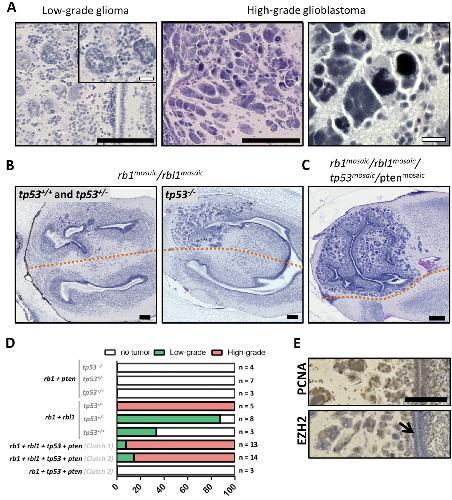
RBL1 (p107) functions as tumor suppressor in glioblastoma and small-cell pancreatic neuroendocrine carcinoma in Xenopus tropicalis.,
Naert T,Dimitrakopoulou D,Tulkens D,Demuynck S,Carron M,Noelanders R,Eeckhout L,Van Isterdael G,Deforce D,Vanhove C,Van Dorpe J,Creytens D,Vleminckx K,Vleminckx K,
Oncogene. March 1, 2020; 39(13):0950-9232.
|
|
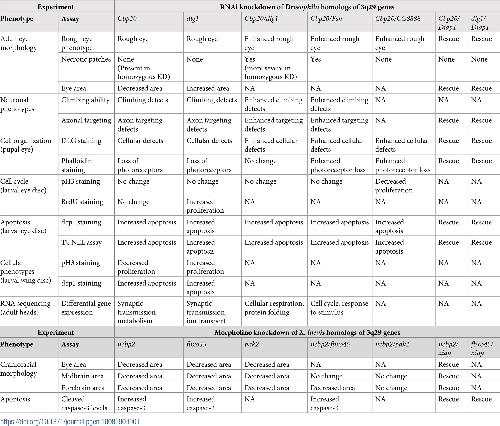
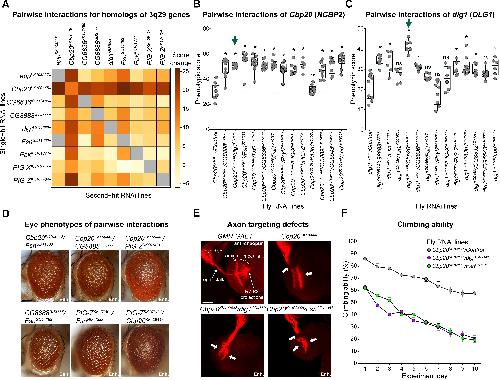
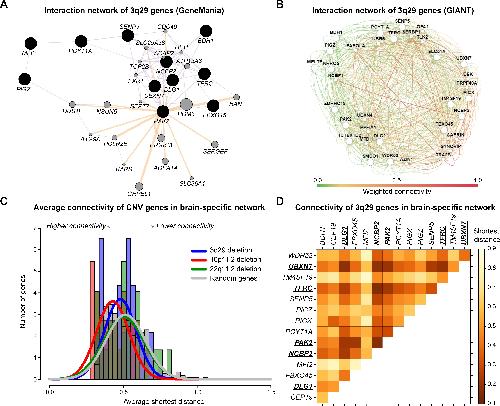
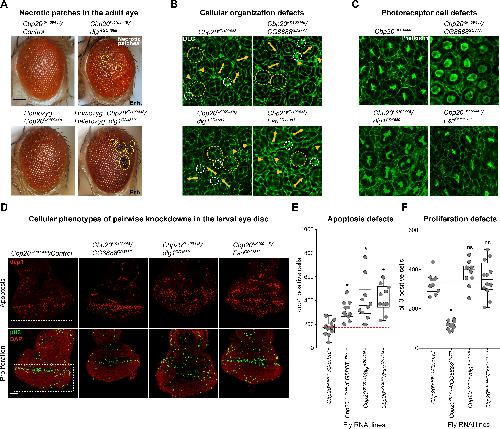
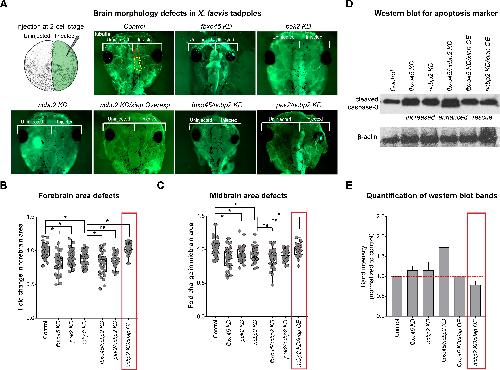
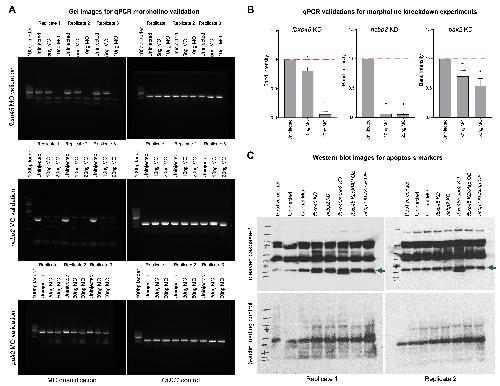
NCBP2 modulates neurodevelopmental defects of the 3q29 deletion in Drosophila and Xenopus laevis models.,
Singh MD,Jensen M,Lasser M,Huber E,Yusuff T,Pizzo L,Lifschutz B,Desai I,Kubina A,Yennawar S,Kim S,Iyer J,Rincon-Limas DE,Lowery LA,Girirajan S,
PLoS Genet. February 13, 2020; 16(2):1553-7404.
|
|
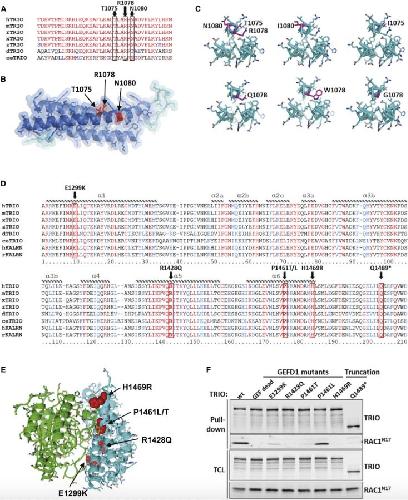
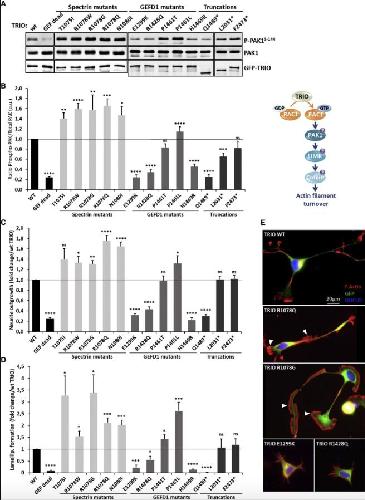
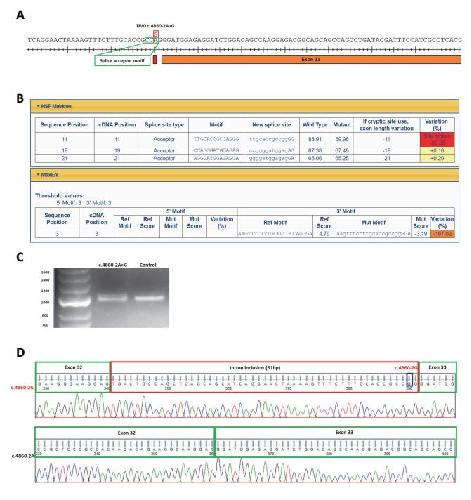
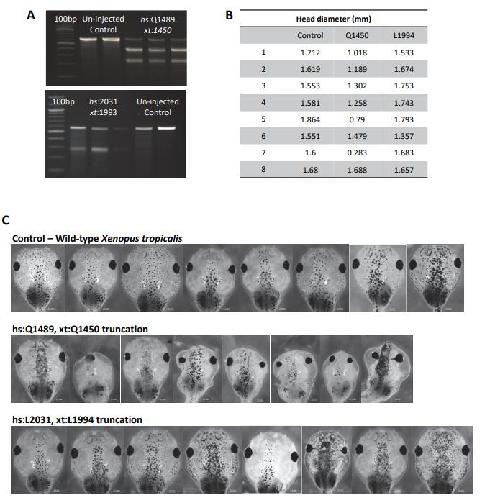
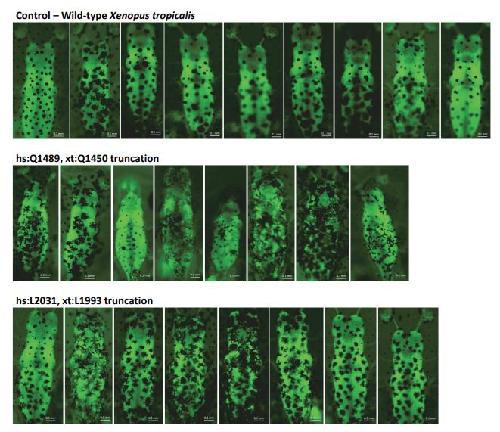
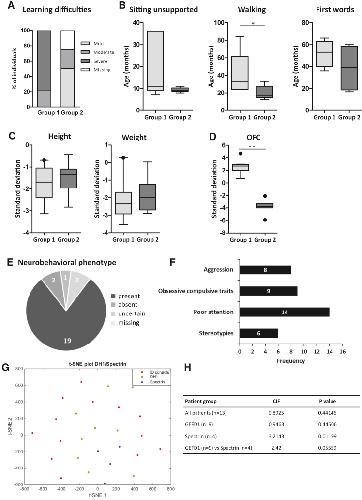
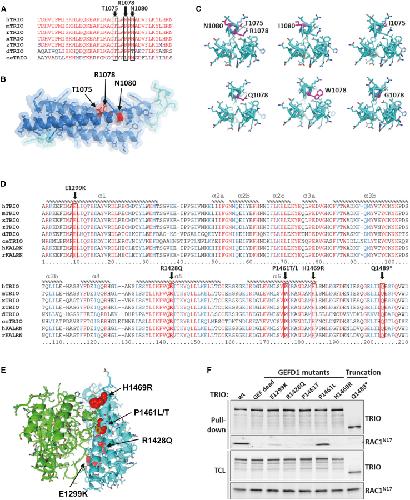
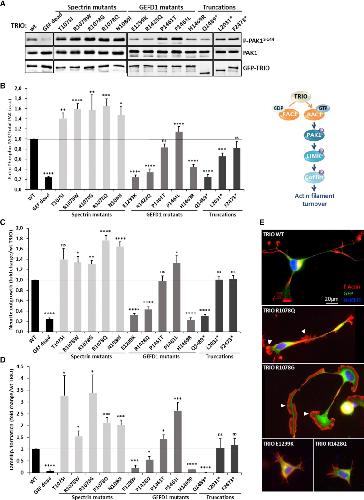
Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders.,
Barbosa S,Greville-Heygate S,Bonnet M,Godwin A,Fagotto-Kaufmann C,Kajava AV,Laouteouet D,Mawby R,Wai HA,Dingemans AJM,Hehir-Kwa J,Willems M,Capri Y,Mehta SG,Cox H,Goudie D,Vansenne F,Turnpenny P,Vincent M,Cogné B,Lesca G,Hertecant J,Rodriguez D,Keren B,Burglen L,Gérard M,Putoux A,Cantagrel V,Siquier-Pernet K,Rio M,Banka S,Sarkar A,Steeves M,Parker M,Clement E,Moutton S,Tran Mau-Them F,Piton A,de Vries BBA,Guille M,Debant A,Schmidt S,Baralle D,
Am J Hum Genet. March 5, 2020; 106(3):1537-6605.
|
|
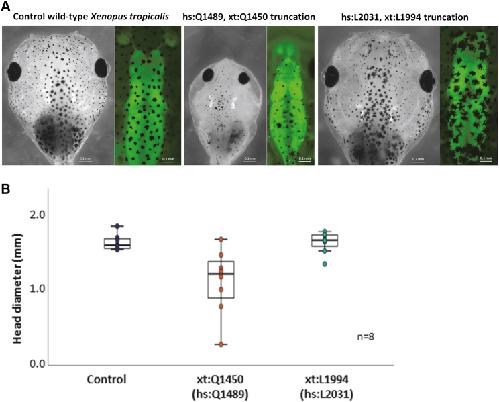
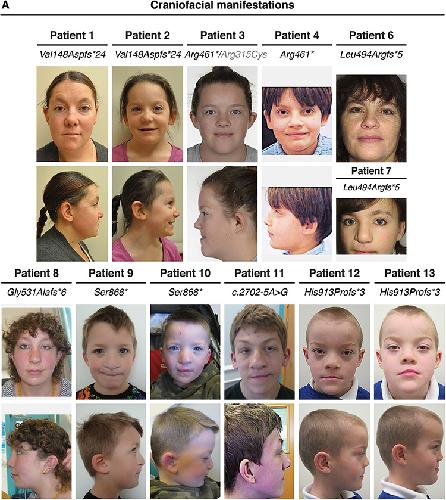
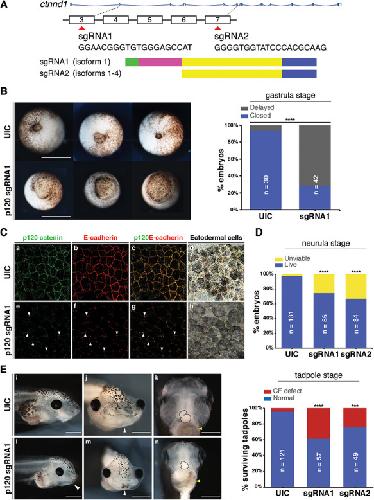
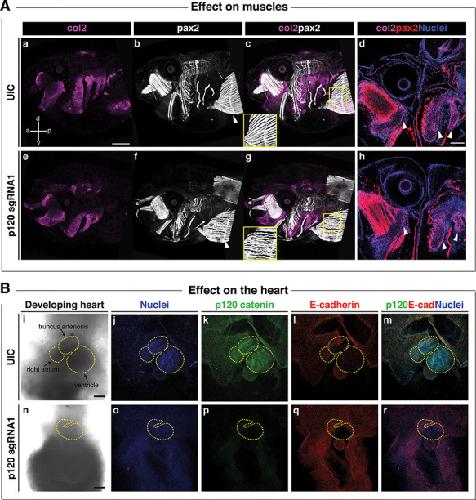
Novel truncating mutations in CTNND1 cause a dominant craniofacial and cardiac syndrome.,
Alharatani R,Ververi A,Beleza-Meireles A,Ji W,Mis E,Patterson QT,Griffin JN,Bhujel N,Chang CA,Dixit A,Konstantino M,Healy C,Hannan S,Neo N,Cash A,Li D,Bhoj E,Zackai EH,Cleaver R,Baralle D,McEntagart M,Newbury-Ecob R,Scott R,Hurst JA,Au PYB,Hosey MT,Khokha M,Marciano DK,Lakhani SA,Liu KJ,Liu KJ,
Hum Mol Genet. July 21, 2020; 29(11):1460-2083.
|
|
A PKD1L3 splice variant in taste buds is not cleaved at the G protein-coupled receptor proteolytic site.,
Kashyap P,Ng C,Wang Z,Li B,Arif Pavel M,Martin H,Yu Y,
Biochem Biophys Res Commun. May 14, 2019; 512(4):1090-2104.
|
|
KCNC1-related disorders: new de novo variants expand the phenotypic spectrum.,
Park J,Koko M,Hedrich UBS,Hermann A,Cremer K,Haberlandt E,Grimmel M,Alhaddad B,Beck-Woedl S,Harrer M,Karall D,Kingelhoefer L,Tzschach A,Matthies LC,Strom TM,Ringelstein EB,Sturm M,Engels H,Wolff M,Lerche H,Haack TB,
Ann Clin Transl Neurol. July 1, 2019; 6(7):2328-9503.
|
|
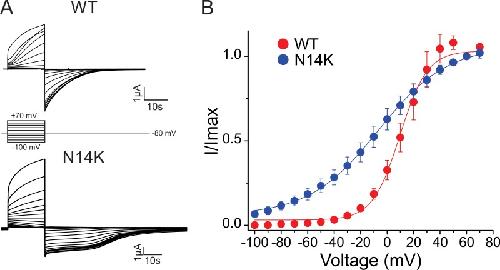
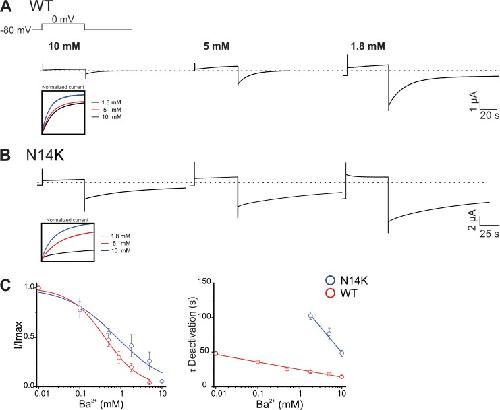
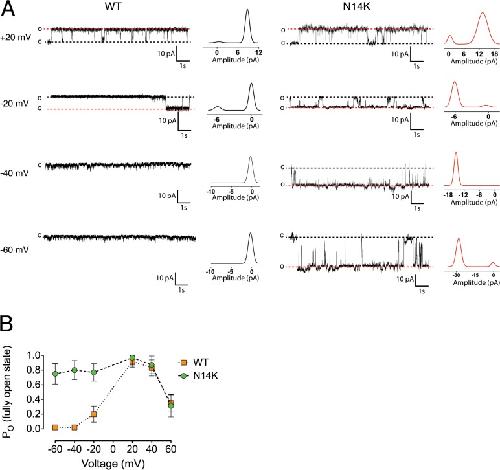
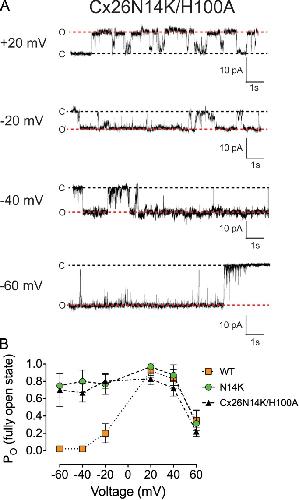
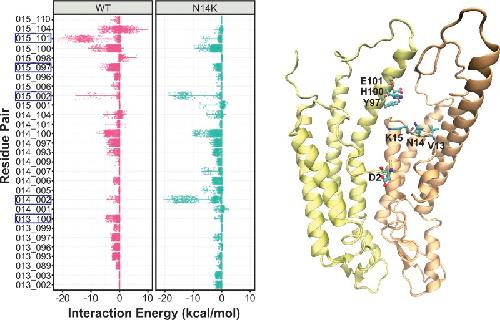
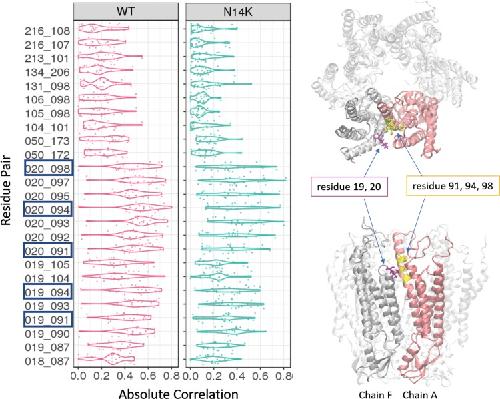
The connexin26 human mutation N14K disrupts cytosolic intersubunit interactions and promotes channel opening.,
Valdez Capuccino JM,Chatterjee P,García IE,Botello-Smith WM,Zhang H,Harris AL,Luo Y,Contreras JE,
J Gen Physiol. March 4, 2019; 151(3):1540-7748.
|
|
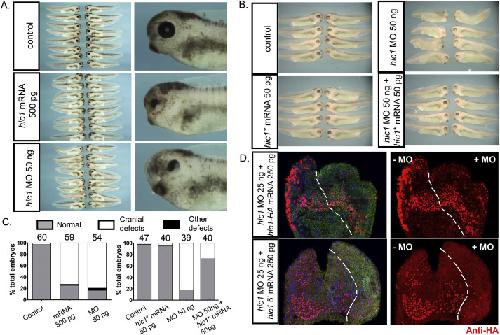
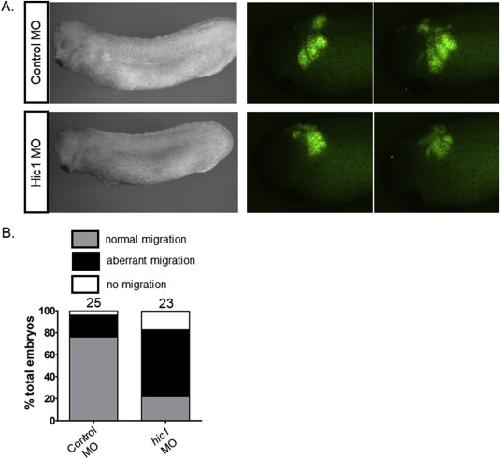
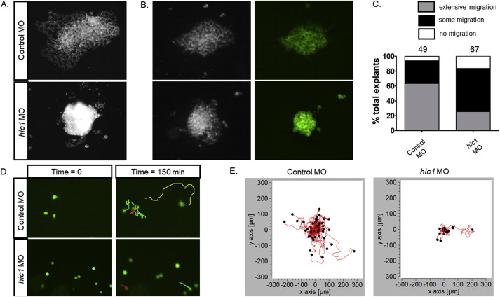
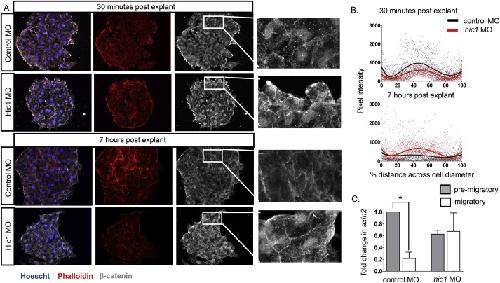
The transcription factor Hypermethylated in Cancer 1 (Hic1) regulates neural crest migration via interaction with Wnt signaling.,
Ray H,Chang C,
Dev Biol. July 15, 2020; 463(2):1095-564X.
|
|
Andersen-Tawil Syndrome Is Associated With Impaired PIP2 Regulation of the Potassium Channel Kir2.1.,
Handklo-Jamal R,Meisel E,Yakubovich D,Vysochek L,Beinart R,Glikson M,McMullen JR,Dascal N,Nof E,Oz S,
Front Pharmacol. April 7, 2020; 11:1663-9812.
|
|
Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav 1.7 channel mutation.,
Rühlmann AH,Körner J,Hausmann R,Bebrivenski N,Neuhof C,Detro-Dassen S,Hautvast P,Benasolo CA,Meents J,Machtens JP,Schmalzing G,Lampert A,
Br J Pharmacol. October 1, 2020; 177(19):1476-5381.
|
|
Establishing embryonic territories in the context of Wnt signaling.,
Velloso I,Maia LA,Amado NG,Reis AH,He X,Abreu JG,
Int J Dev Biol. January 1, 2021; 65(4-5-6):1696-3547.
|
|
Characterization of the GABRB2-Associated Neurodevelopmental Disorders.,
El Achkar CM,Harrer M,Smith L,Kelly M,Iqbal S,Maljevic S,Niturad CE,Vissers LELM,Poduri A,Yang E,Lal D,Lerche H,Møller RS,Olson HE,
Ann Neurol. March 1, 2021; 89(3):1531-8249.
|
| Editorial: Xenopus Models of Organogenesis and Disease., Griffin JN,Liu KJ,Liu KJ,Sempou E, Front Physiol. January 1, 2020; 11:1664-042X. |
|
Molecular Mechanism of Autosomal Recessive Long QT-Syndrome 1 without Deafness.,
Oertli A,Rinné S,Moss R,Kääb S,Seemann G,Beckmann BM,Decher N,
Int J Mol Sci. January 23, 2021; 22(3):1422-0067.
|
|
Aquatic models of human ciliary diseases.,
Corkins ME,Krneta-Stankic V,Kloc M,Miller RK,
Genesis. February 1, 2021; 59(1-2):1526-968X.
|
|
Sleep-related hypermotor epilepsy associated mutations uncover important kinetic roles of α4β2- nicotinic acetylcholine receptor intracellular structures.,
Weltzin MM,George AA,Lukas RJ,Whiteaker P,
PLoS One. March 3, 2021; 16(3):1932-6203.
|
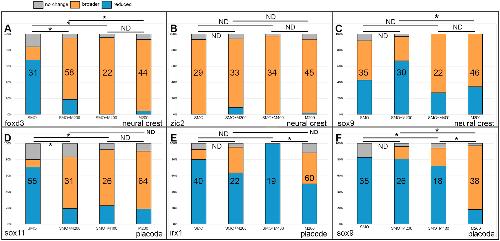
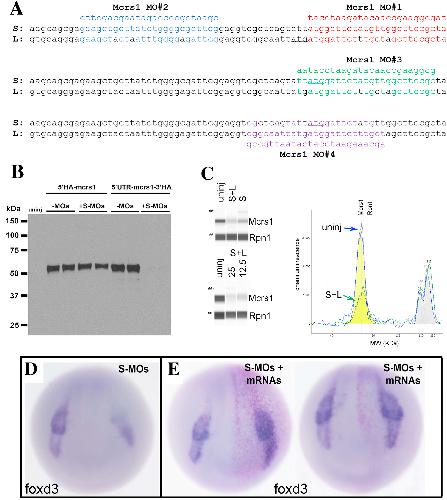
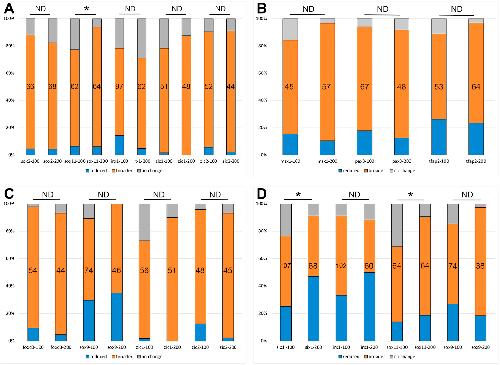
|
Mcrs1 interacts with Six1 to influence early craniofacial and otic development.,
Neilson KM,Keer S,Bousquet N,Macrorie O,Majumdar HD,Kenyon KL,Alfandari D,Alfandari D,Moody SA,
Dev Biol. November 1, 2020; 467(1-2):1095-564X.
|
| Cation leak underlies neuronal excitability in an HCN1 developmental and epileptic encephalopathy., Bleakley LE,McKenzie CE,Soh MS,Forster IC,Pinares-Garcia P,Sedo A,Kathirvel A,Churilov L,Jancovski N,Maljevic S,Berkovic SF,Scheffer IE,Petrou S,Santoro B,Reid CA, Brain. August 17, 2021; 144(7):1460-2156. |
|
Dissecting the Molecular Determinants of GABAA Receptors Current Rundown, a Hallmark of Refractory Human Epilepsy.,
Cifelli P,Di Angelantonio S,Alfano V,Morano A,De Felice E,Aronica E,Ruffolo G,Palma E,
Brain Sci. March 30, 2021; 11(4):2076-3425.
|
|
Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer.,
Levin M,
Cell. April 15, 2021; :1097-4172.
|
| Modeling endoderm development and disease in Xenopus., Edwards NA,Zorn AM, Curr Top Dev Biol. January 1, 2021; 145:1557-8933. |
|
A polycystin-2 protein with modified channel properties leads to an increased diameter of renal tubules and to renal cysts.,
Grosch M,Brunner K,Ilyaskin AV,Schober M,Staudner T,Schmied D,Stumpp T,Schmidt KN,Madej MG,Pessoa TD,Othmen H,Kubitza M,Osten L,de Vries U,Mair MM,Somlo S,Moser M,Kunzelmann K,Ziegler C,Haerteis S,Korbmacher C,Witzgall R,
J Cell Sci. August 15, 2021; 134(16):1477-9137.
|
|
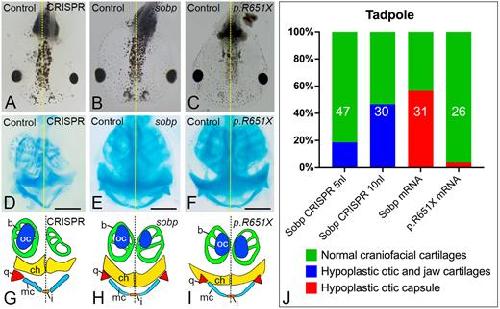
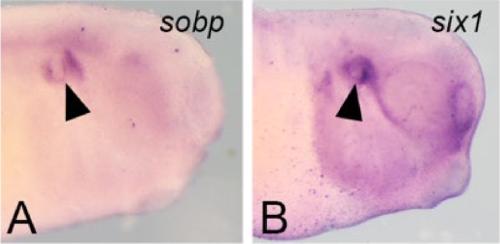
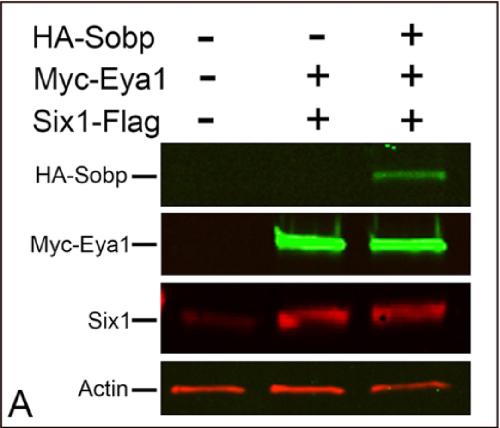
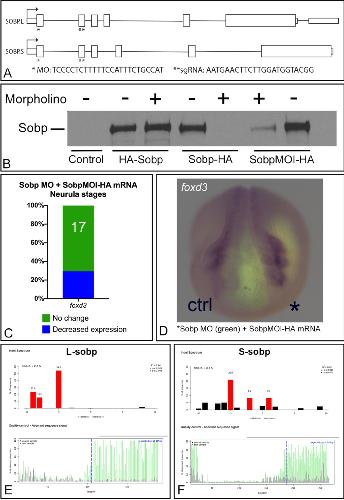
Sobp modulates the transcriptional activation of Six1 target genes and is required during craniofacial development.,
Tavares ALP,Jourdeuil K,Neilson KM,Majumdar HD,Moody SA,
Development. September 1, 2021; 148(17):1477-9129.
|
|
Comparative analysis of alternating hemiplegia of childhood and rapid-onset dystonia-parkinsonism ATP1A3 mutations reveals functional deficits, which do not correlate with disease severity.,
Lazarov E,Hillebrand M,Schröder S,Ternka K,Hofhuis J,Ohlenbusch A,Barrantes-Freer A,Pardo LA,Fruergaard MU,Nissen P,Brockmann K,Gärtner J,Rosewich H,
Neurobiol Dis. September 1, 2020; 143:1095-953X.
|
|
Function of chromatin modifier Hmgn1 during neural crest and craniofacial development.,
Ihewulezi C,Saint-Jeannet JP,
Genesis. October 1, 2021; 59(10):1526-968X.
|
|
Modeling human congenital disorders with neural crest developmental defects using patient-derived induced pluripotent stem cells.,
Okuno H,Okano H,
Regen Ther. August 24, 2021; 18:2352-3204.
|
|
The Role of RNA-Binding Proteins in Vertebrate Neural Crest and Craniofacial Development.,
Forman TE,Dennison BJC,Fantauzzo KA,
J Dev Biol. August 27, 2021; 9(3):2221-3759.
|
|
Physical basis for distinct basal and mechanically gated activity of the human K+ channel TRAAK.,
Rietmeijer RA,Sorum B,Li B,Brohawn SG,
Neuron. September 15, 2021; 109(18):0896-6273.
|
|
Structural comparison of GLUT1 to GLUT3 reveal transport regulation mechanism in sugar porter family.,
Custódio TF,Paulsen PA,Frain KM,Pedersen BP,
Life Sci Alliance. February 3, 2021; 4(4):2575-1077.
|
|
Generation of a new six1-null line in Xenopus tropicalis for study of development and congenital disease.,
Coppenrath K,Tavares ALP,Shaidani NI,Wlizla M,Moody SA,Horb M,
Genesis. December 1, 2021; 59(12):1526-968X.
|
|
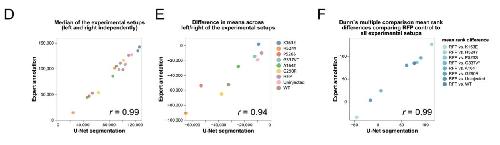
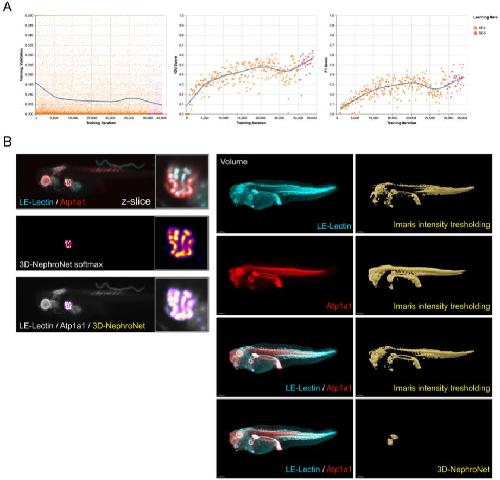
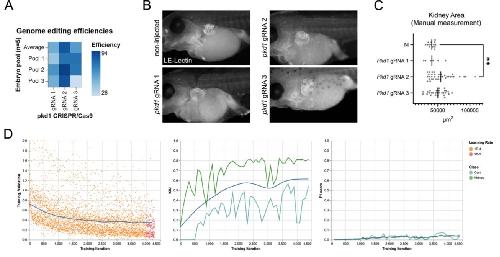
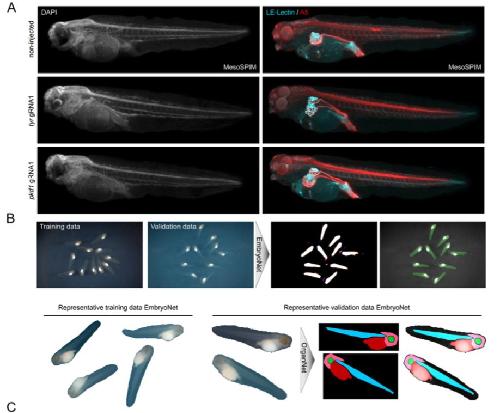
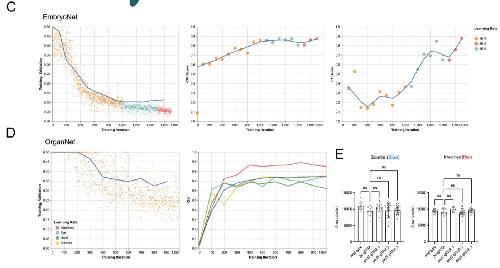
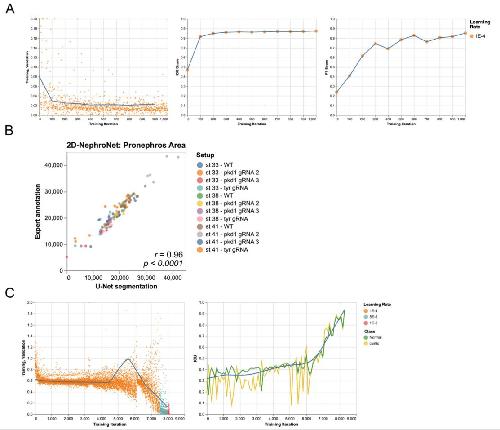
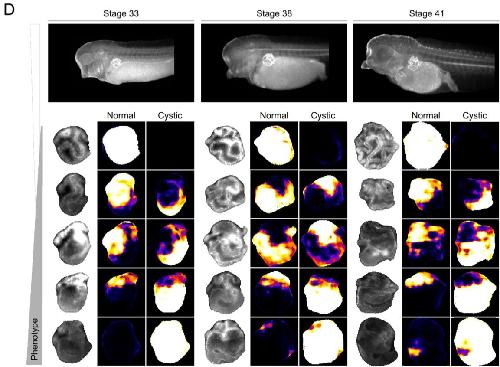
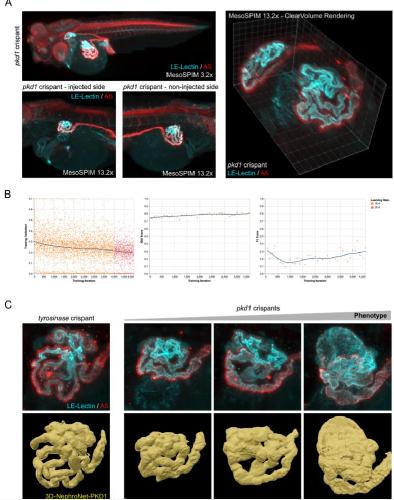
Deep learning is widely applicable to phenotyping embryonic development and disease.,
Naert T,Çiçek Ö,Ogar P,Bürgi M,Shaidani NI,Kaminski MM,Xu Y,Xu Y,Grand K,Vujanovic M,Prata D,Hildebrandt F,Brox T,Ronneberger O,Voigt FF,Helmchen F,Loffing J,Horb ME,Willsey HR,Lienkamp SS,
Development. November 1, 2021; 148(21):1477-9129.
|
|
Functional cross-talk between phosphorylation and disease-causing mutations in the cardiac sodium channel Nav1.5.,
Galleano I,Harms H,Choudhury K,Khoo K,Delemotte L,Pless SA,
Proc Natl Acad Sci U S A. August 17, 2021; 118(33):1091-6490.
|
|
Reduced Retinoic Acid Signaling During Gastrulation Induces Developmental Microcephaly.,
Gur M,Bendelac-Kapon L,Shabtai Y,Pillemer G,Fainsod A,
Front Cell Dev Biol. January 1, 2022; 10:2296-634X.
|
|
Thyroid Hormone Receptor α Controls the Hind Limb Metamorphosis by Regulating Cell Proliferation and Wnt Signaling Pathways in Xenopus tropicalis.,
Tanizaki Y,Shibata Y,Zhang H,Shi YB,Shi YB,
Int J Mol Sci. January 22, 2022; 23(3):1422-0067.
|
|
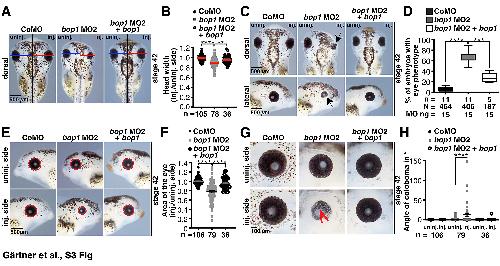
Functions of block of proliferation 1 during anterior development in Xenopus laevis.,
Gärtner C,Meßmer A,Dietmann P,Kühl M,Kühl SJ,
PLoS One. August 2, 2022; 17(8):1932-6203.
|
|
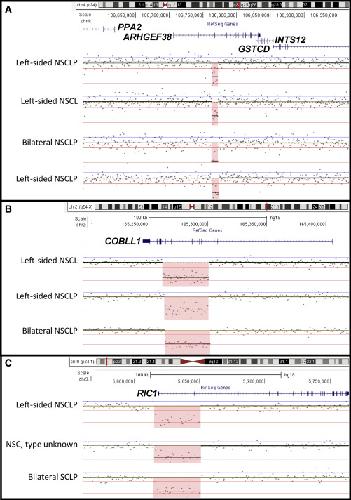
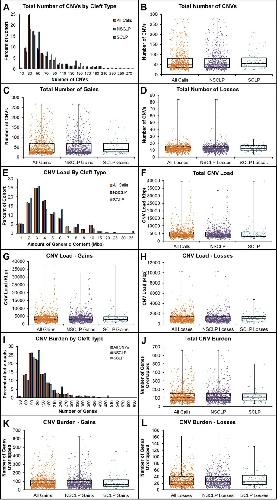
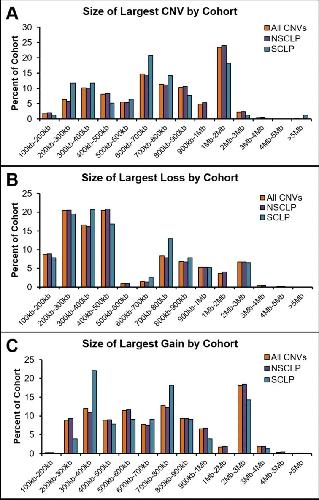
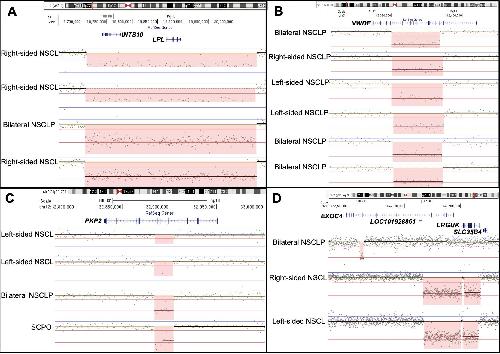
Genome-wide analysis of copy-number variation in humans with cleft lip and/or cleft palate identifies COBLL1, RIC1, and ARHGEF38 as clefting genes.,
Lansdon LA,Dickinson A,Arlis S,Liu H,Hlas A,Hahn A,Bonde G,Long A,Standley J,Tyryshkina A,Wehby G,Lee NR,Daack-Hirsch S,Mohlke K,Girirajan S,Darbro BW,Cornell RA,Houston DW,Murray JC,Manak JR,
Am J Hum Genet. January 5, 2023; 110(1):1537-6605.
|
|
Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants.,
Rinné S,Oertli A,Nagel C,Tomsits P,Jenewein T,Kääb S,Kauferstein S,Loewe A,Beckmann BM,Decher N,
Int J Mol Sci. January 10, 2023; 24(2):1422-0067.
|
|
Phenotype-genotype relationships in Xenopus sox9 crispants provide insights into campomelic dysplasia and vertebrate jaw evolution.,
Hossain N,Igawa T,Suzuki M,Suzuki M,Tazawa I,Nakao Y,Hayashi T,Suzuki N,Ogino H,
Dev Growth Differ. October 1, 2023; 65(8):1440-169X.
|
|
Inactivation influences the extent of inhibition of voltage-gated Ca+2 channels by Gem-implications for channelopathies.,
Allam S,Levenson-Palmer R,Chia Chang Z,Kaur S,Cernuda B,Raman A,Booth A,Dobbins S,Suppa G,Yang J,Buraei Z,
Front Physiol. January 1, 2023; 14:1664-042X.
|
|
Time-resolved quantitative proteomic analysis of the developing Xenopus otic vesicle reveals putative congenital hearing loss candidates.,
Baxi AB,Nemes P,Moody SA,
iScience. September 15, 2023; 26(9):2589-0042.
|
|
Advancements in the use of xenopus oocytes for modelling neurological disease for novel drug discovery.,
O'Connor EC,Kambara K,Bertrand D,
Expert Opin Drug Discov. February 1, 2024; 19(2):1746-045X.
|
|
FGFR1 variants contributed to families with tooth agenesis.,
Yao S,Zhou X,Zhou X,Gu M,Zhang C,Bartsch O,Vona B,Fan L,Ma L,Pan Y,
Hum Genomics. October 13, 2023; 17(1):1479-7364.
|
|
Using Xenopus to discover new candidate genes involved in BOR and other congenital hearing loss syndromes.,
Neal SJ,Rajasekaran A,Jusić N,Taylor L,Read M,Alfandari D,Alfandari D,Pignoni F,Moody SA,
J Exp Zool B Mol Dev Evol. October 13, 2023; :1552-5015.
|
|
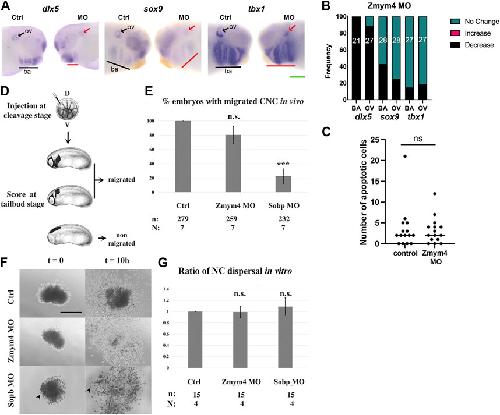
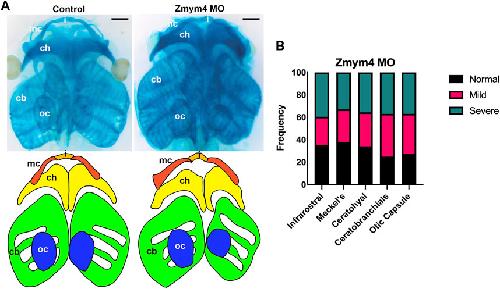
Zmym4 is required for early cranial gene expression and craniofacial cartilage formation.,
Jourdeuil K,Neilson KM,Cousin H,Tavares ALP,Majumdar HD,Alfandari D,Alfandari D,Moody SA,
Front Cell Dev Biol. January 1, 2023; 11:2296-634X.
|
| Remarkable effect of transdermal nicotine in children with CHRNA4-related autosomal dominant sleep-related hypermotor epilepsy., Lossius K,de Saint Martin A,Myren-Svelstad S,Bjørnvold M,Minken G,Seegmuller C,Valenti Hirsch MP,Chelly J,Steinlein O,Picard F,Brodtkorb E, Epilepsy Behav. April 1, 2020; 105:1525-5069. |
| Functional and clinical characterization of a novel homozygous KCNH2 missense variant in the pore region of Kv11.1 leading to a viable but severe long-QT syndrome., Delinière A,Jaupart L,Janin A,Millat G,Boulin T,Andrini O,Chevalier P, Gene. March 1, 2024; 897:1879-0038. |
| A novel SMARCC1 BAFopathy implicates neural progenitor epigenetic dysregulation in human hydrocephalus., Singh AK,Allington G,Viviano S,McGee S,Kiziltug E,Ma S,Zhao S,Mekbib KY,Shohfi JP,Duy PQ,DeSpenza T,Furey CG,Reeves BC,Smith H,Sousa AMM,Cherskov A,Allocco A,Nelson-Williams C,Haider S,Rizvi SRA,Alper SL,Sestan N,Shimelis H,Walsh LK,Lifton RP,Moreno-De-Luca A,Jin SC,Kruszka P,Deniz E,Kahle KT, Brain. April 4, 2024; 147(4):1460-2156. |
| Genetic models of fibrillinopathies., Summers KM, Genetics. January 3, 2024; 226(1):1943-2631. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}